Mega-scale experimental analysis of protein folding stability in biology and design
science-easyview
생물학 및 설계 분야의 단백질 폴딩 안정성에 대한 대규모 실험 분석
Abstract
DNA 시퀀싱과 머신 러닝의 발전은 엄청난 규모의 단백질 서열과 구조에 대한 통찰력을 제공합니다. 그러나 폴딩을 주도하는 에너지는 구조에서 보이지 않으며 대부분 알려지지 않은 상태로 남아 있습니다. 폴딩의 숨겨진 열역학은 질병을 유발하고, 단백질 진화를 형성하고, 단백질 공학을 안내할 수 있으며, 모든 서열과 구조에 대해 이러한 열역학을 밝히기 위해서는 새로운 접근법이 필요합니다.
이 연구에서는 1주일 실험에서 최대 900,000개의 단백질 도메인에 대한 열역학적 폴딩 안정성을 측정하는 방법인 cDNA 디스플레이 단백질 분해를 제시합니다. 총 180만 건의 측정에서 우리는 모든 단일 아미노산 변이체와 길이가 40–72 아미노산 길이인 331개의 천연 및 148개의 de novo 디자인 단백질 도메인의 선택된 이중 돌연변이를 포괄하는 약 776,000개의 고품질 폴딩 안정성 세트를 선별했습니다. 이 광범위한 데이터 세트를 사용하여 (1) 아미노산 적합성에 영향을 미치는 환경 요인, (2) 단백질 부위 간의 열역학적 결합 (예기치 않은 상호 작용 포함), (3) 진화론적 아미노산 사용과 단백질 폴딩 안정성 사이의 글로벌 차이를 정량화했습니다.
우리는 또한 우리의 접근 방식이 디자인된 단백질에서 안정성 결정 요인을 식별하고 디자인 방법을 평가할 수 있는 방법을 조사했습니다. cDNA 디스플레이 단백질 분해 방법은 빠르고 정확하며 고유하게 확장 가능하며 아미노산 서열이 폴딩 안정성을 인코딩하는 방법에 대한 정량적 규칙을 밝힐 것입니다.
Figure
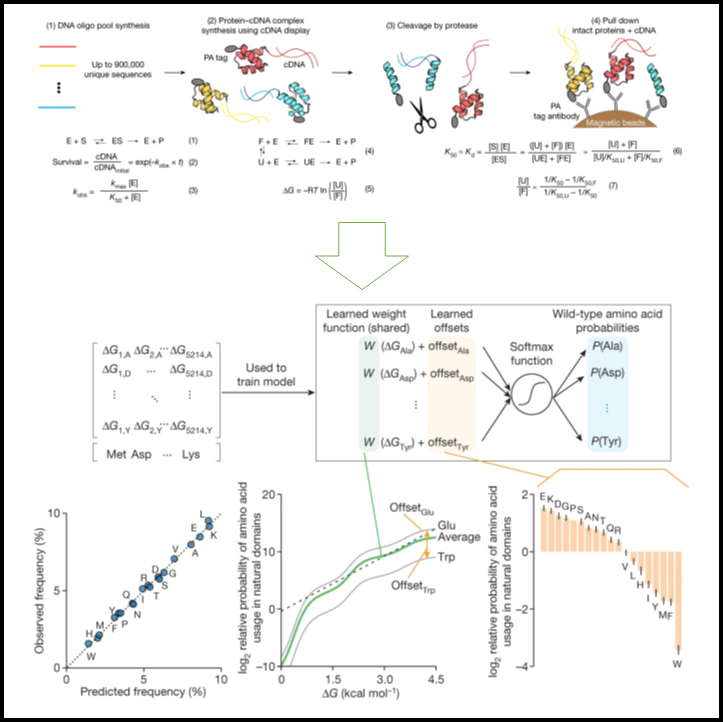
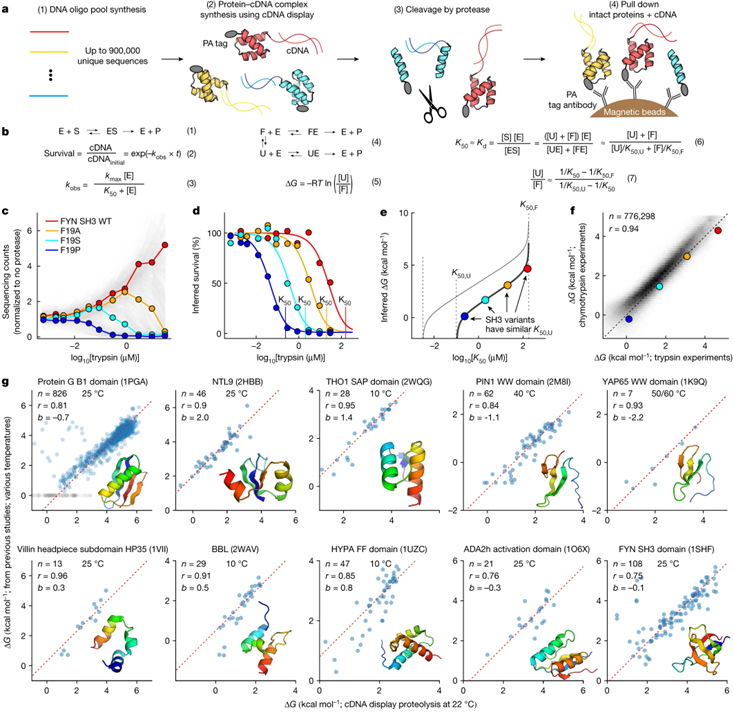
[Figure 1] cDNA 디스플레이를 통한 단백질 폴딩 안정성의 대규모 병렬 측정 (A) DNA 올리고뉴클레오티드 (oligo) 라이브러리를 무세포 cDNA 디스플레이를 사용하여 발현되어 N-말단 14-아미노산 PA 태그와 cDNA에 대한 C-말단 공유 결합이 있는 단백질 생성 후 정량화 과정. (B) 단백질 분해의 열역학적 모델. (1) 프로테아제 효소 (E)와 단백질 기질 (S)은 ES 복합체를 형성하여 절단된 단백질 생성물 (P)을 생성. 절단을 단일 회전 동역학 (3)에 따라 1차 반응 (2)로 모델링. (4) 단백질은 일반적으로 펼쳐진 (U) 상태에서 절단되지만 PA 태그를 절단하여 접힌 (F) 상태에서도 절단 가능. 각 서열의 측정된 K50, 펼쳐진 상태에 대한 예측된 서열 특정 K50 (K50,U) 및 접힌 상태에 대한 범용 K50 (K50,F)을 사용하여 ΔG 결정. (C) 각 인간 FYN SH3 변이체 서열은 사전 선택 라이브러리를 기준으로 시퀀싱 카운트 추적. (D) 상이한 프로테아제 농도에서 c로부터의 4개의 서열에 대한 절단되지 않은 단백질의 추정된 생존. (E) K50,U의 다른 값에 대한 K50과 ΔG 사이의 관계. (F) 품질 필터 (데이터 세트 2) 후 독립적인 트립신과 키모트립신 실험 간의 ΔG 추정치의 일관성. (G) 높은 처리량 ΔG 측정은 표시된 도메인의 돌연변이에 대한 정제된 샘플의 게시된 데이터와 일치.
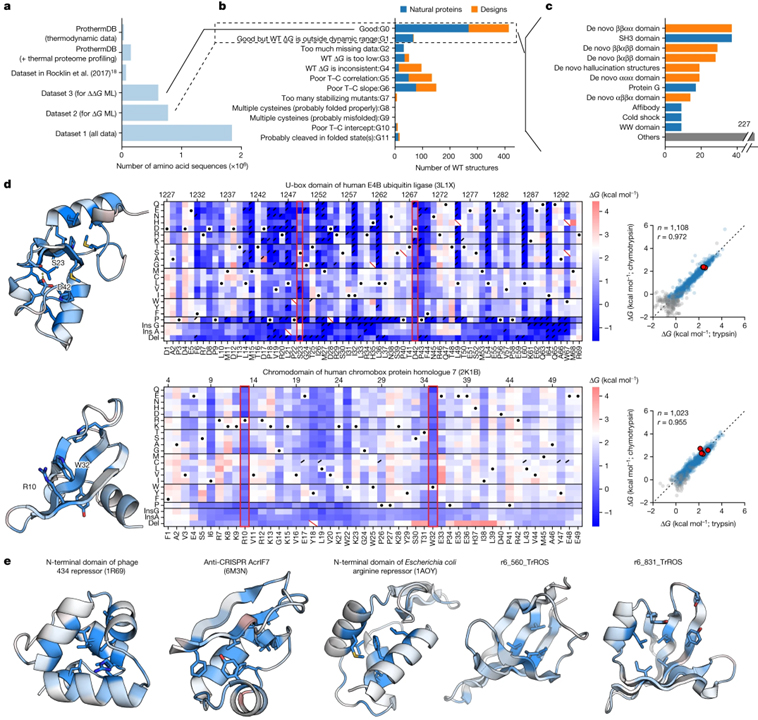
[Figure 2] 설계된 단백질과 천연 단백질의 안정성에 대한 포괄적인 돌연변이 분석 (A) 데이터세트 1, 2, 3과 비교한 기존 데이터세트의 크기. (B) 각 야생형 (WT) 서열에 대한 돌연변이 스캐닝 결과의 분류. (C) 도메인 패밀리로 그룹화된 b에서 G0으로 분류된 야생형 구조. (D) 인간 E4B 유비퀴틴 리가아제 (PDB ID: 3L1X)의 U-box 도메인 (상단) 및 크로모박스 단백질 상동체 7 (PDB ID: 2K1B) (하단)의 크로모 도메인에 대한 돌연변이 스캐닝 결과. (E) d와 같이 제시된 데이터 세트의 다른 5개 도메인 구조.
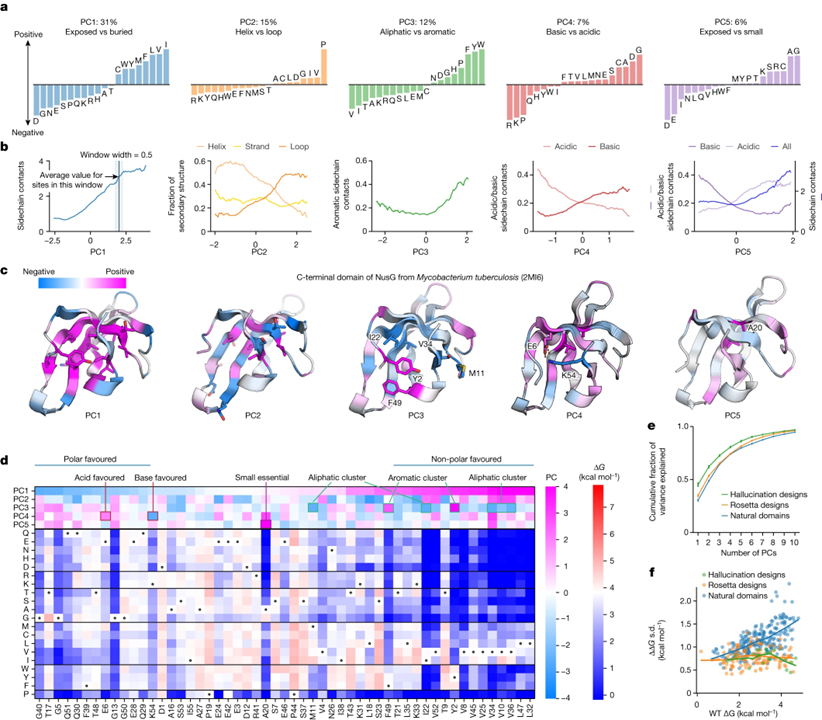
[Figure 3] 위치에서 아미노산 안정성을 결정하는 환경 요인 (A) 아미노산이 한 부위에서 안정화 또는 불안정화되는 지배적인 경향을 보여주는 안정성 데이터의 주성분 (PC1–PC5). (B) 모든 17,093개 사이트에 대한 주성분 값 (x축)과 모델링된 3D 구조 (y축)에서 해당 사이트의 환경 속성 간의 관계. (C) PC1-PC5의 값으로 채색된 사이트가 있는 NusG (PDB ID: 2MI6)의 C-말단 도메인의 구조. (D) 각 사이트에서 PC1의 값으로 정렬된 NusG에 대한 돌연변이 스캐닝 결과. (E) 천연 도메인 (파란색), Rosetta 디자인 (주황색), 환각 디자인 (녹색)의 개별 주성분 분석을 위해 주성분으로 설명된 총 분산의 누적 비율. (F) 각 사이트의 ΔΔG 데이터에서 야생형 안정성과 분산 (sd) 사이의 관계.
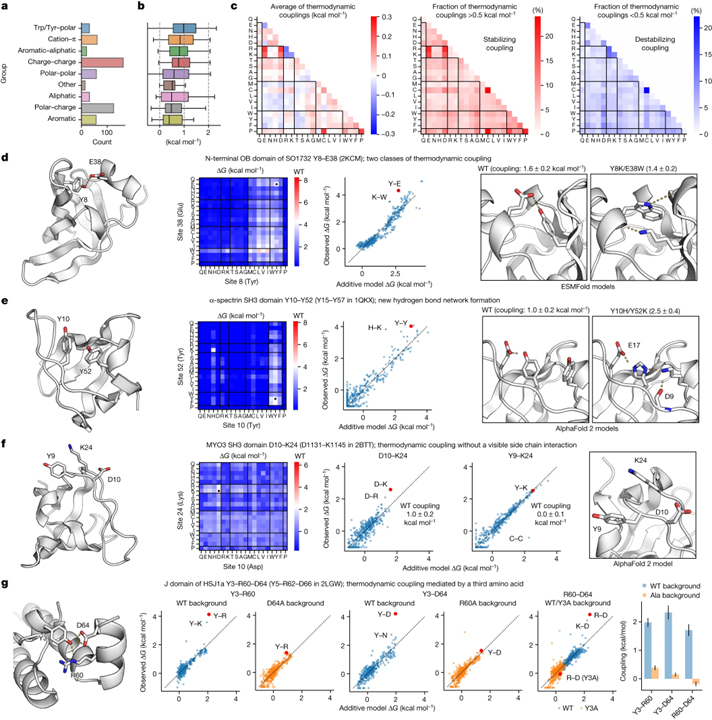
[Figure 4] 아미노산 쌍 사이의 열역학적 결합의 정량화 (A-B) 철저한 이중 돌연변이 분석을 위해 선택된 190개 도메인에서 559쌍의 아미노산의 분류 (총 210,000 ΔG 측정) (a)와 당사의 첨가제 모델에 따른 야생형 아미노산 쌍의 열역학적 결합 (b). (C) 평균 열역학적 결합 (왼쪽) 및 모든 아미노산 조합 (야생형 및 돌연변이)에 대해 0.5kcal mol-1보다 강한 양성 (중간) 또는 음성 (오른쪽) 결합을 갖는 쌍의 분율. (D-E) SO1732 (PDB ID: 2KCM) Y8–E38의 N-말단 OB-도메인에 대한 열역학적 결합 (d) 및 α-스펙트린 SH3 도메인 (PDB ID: 1QKX) Y10–Y52 (PDB 구조의 Y15–Y57) (e). (F) MYO3 SH3 도메인 D10-K24 (PDB ID 2BTT의 D1131-K1145)에서 눈에 보이는 측쇄 상호작용이 없는 열역학적 결합. (G) HSJ1a의 J 도메인에 있는 세 번째 아미노산에 의해 매개되는 열역학적 결합.
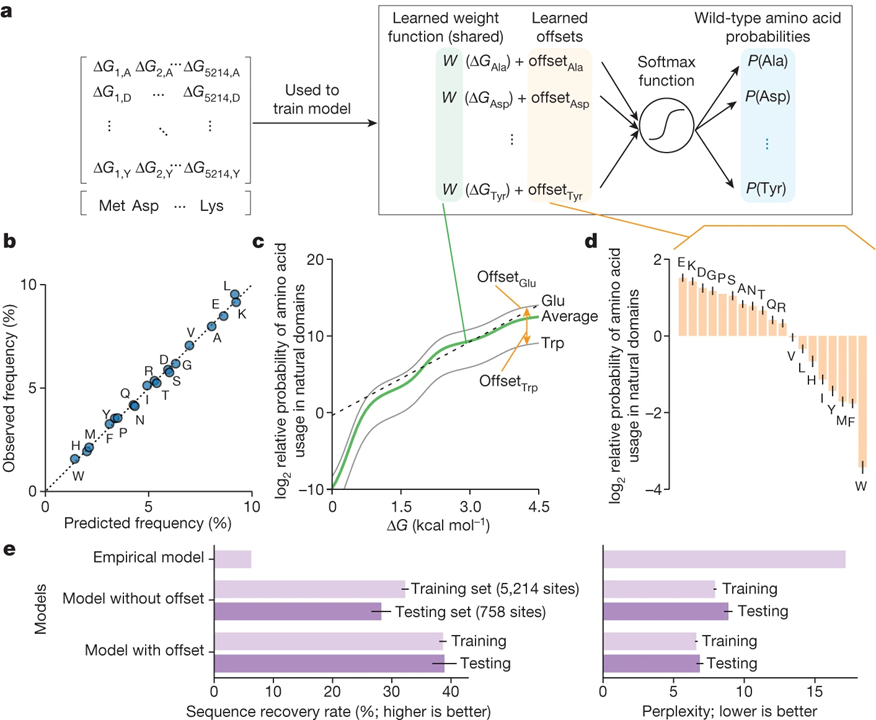
[Figure 5] 천연 단백질의 아미노산 빈도에 의한 체계적인 안정성 최대화 변화 (A) 각 단백질 변이체의 폴딩 안정성 (ΔG)으로부터 야생형 아미노산을 예측하기 위한 분류기 모델. (B) 피팅 후 분류기 모델에 따라 예측되고 관찰된 아미노산 빈도. (C) 피팅 후 분류기 모델의 가중치 함수. (D) 피팅 후 분류자 모델에서 19개 아미노산에 대한 상대적 오프셋. (E) 여러 모델을 사용하여 야생형 아미노산을 예측하기 위한 서열 회수율 (왼쪽) 및 당혹도 (오른쪽).
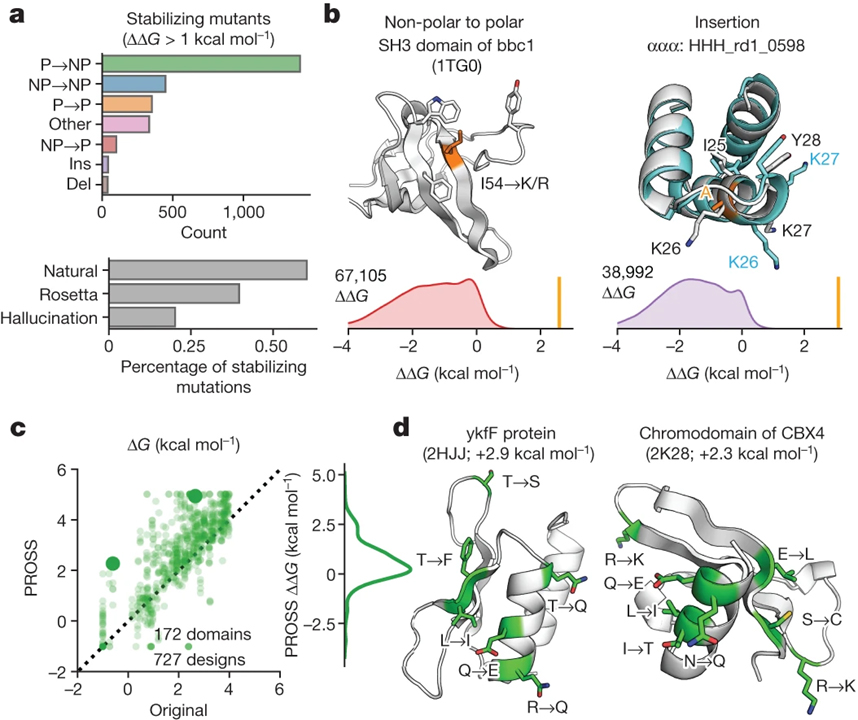
[Figure 6] 대규모 데이터를 단백질 설계에 적용 (A) 돌연변이 유형별로 세분화된 천연 도메인, Rosetta 디자인 및 환각 디자인에서 발견되는 안정화 돌연변이. (B) 돌연변이 유형에 대한 ΔΔG 값의 분포와 함께 우리의 분석에서 발견된 안정화 돌연변이의 두 가지 예. (C) PROSS44에 의한 재설계 전 (x축) 및 후 (y축) 테스트 도메인의 안정성. (D) PROSS에 의해 재설계된 도메인의 AlphaFold 모델.
Disscussion
cDNA 디스플레이 단백질 분해 방법은 폴딩 안정성 실험의 규모를 크게 확장합니다. 그럼에도 불구하고 이 방법에는 현재 주목할만한 한계가 있습니다. 첫째, 우리의 분석은 세포가 없는 환경에서 발현하고 접히는 단백질로 제한되며 cDNA 디스플레이와 호환됩니다. 각 라이브러리 구성의 농도가 매우 낮기 때문에 복합체에서 안정화된 단백질은 이 분석에 적합하지 않을 수 있습니다. 둘째, 천연 조건에서 단백질을 분해하기 때문에 추론된 열역학적 안정성은 (1) 폴딩이 완전히 협력적일 때 (풀림 없이 세그먼트가 절단되지 않음), (2) 분석 중 폴딩이 평형 상태에 있을 때 (응집으로 인해 동역학 안정성 또는 거짓 안정성 없음), (3) K50,U가 정확하게 추론될 때, (4) 절단으로 인해 cDNA의 해리 (C-말단을 유지할 수 있는 최소한의 이황화물 또는 기타 가교결합)일 때만 정확합니다. (5) 분해율은 현재 동적 범위를 약 5kcal/mol로 제한하는 분석의 측정 가능한 범위 내에 속합니다. 많은 도메인, 특히 더 큰 단백질 구조는 이러한 조건을 충족하지 않습니다. 우리의 돌연변이 스캐닝 데이터는 종종 비협조성 또는 응집의 사례를 제안했지만, 이러한 잠재적 인공물은 돌연변이 스캐닝 없이 개별 서열을 분석할 때 보이지 않을 수 있습니다. 또한 두 프로테아제가 동등하게 영향을 받고 절단이 개별 돌연변이에 지나치게 민감하지 않은 경우 접힌 상태로부터의 절단은 돌연변이 스캐닝 데이터로도 감지할 수 없습니다. cDNA 디스플레이 단백질 분해와 화학적 변성 (펄스 단백질 분해)을 결합하면 이러한 장애물 중 일부를 극복하고 덜 협력적이거나 더 높은 안정성을 가진 단백질의 대규모 분석을 가능하게 할 수 있습니다. DNA 합성의 발전은 또한 여기에서 연구된 가장 큰 도메인 (72개 아미노산) 이상으로 cDNA 디스플레이 단백질 분해를 확장하는 것을 가능하게 할 것입니다. 마지막으로 다중 측정 및 자동화된 데이터 처리는 신뢰할 수 없는 데이터를 제외하기 위해 노력했지만 부정확성을 가져올 가능성이 있습니다. 주목할만한 개별 결과의 경우 원시 데이터를 검토하는 것이 도움이 될 수 있으며 모든 데이터와 코드를 포함하여 모든 피팅을 재생성했습니다.
이러한 한계에도 불구하고, cDNA 디스플레이 단백질 분해의 독특한 규모는 단백질 안정성 연구를 위한 완전히 새로운 가능성을 열어줍니다. Protein Data Bank의 거의 모든 작은 구조에서 단일 돌연변이를 종합적으로 측정하여 다양한 위치에서의 아미노산 적합성 경향, 단일 및 이중 돌연변이의 영향 경향, 안정성이 서열 진화에 미치는 영향에 대한 경향 등 몇 가지 글로벌 경향을 정량화했습니다. 이러한 추세와 함께 우리의 분석은 소규모 방법으로 식별하기 어려운 수백 가지의 예외적인 사례를 발견했습니다. 여기에는 극단적인 효과가 있는 돌연변이, 비정상적인 안정성 환경이 있는 사이트, 비정상적으로 강한 열역학적 결합과의 쌍 상호 작용이 포함됩니다. 증착된 NMR 구조에는 존재하지 않는 인간 HSJ1a의 J 도메인에서 식별한 열역학적 결합은 대규모 안정성 분석이 솔루션의 구조적 세부 사항을 밝히기 위한 다른 방법을 보완할 수 있는 방법을 강조합니다. 안정성을 연구하는 것 외에도 cDNA 디스플레이 단백질 분해는 설계 방법을 체계적으로 개선하고 폴딩 안정성과 기능 사이의 관계를 분석하기 위해 대규모로 설계된 단백질을 분석하는 것을 포함하여 다른 응용 분야를 가질 것입니다.
단백질 안정성과 서열 의존성에 대한 정확하고 정량적인 이해를 달성하는 것은 오랫동안 생물 물리학의 중심 목표였습니다. 우리는 수백만 개의 cDNA 디스플레이 단백질 분해 측정이 절대 폴딩 안정성과 돌연변이의 영향을 예측하는 차세대 딥 러닝 모델의 기초를 형성하는 것을 상상합니다. 딥 러닝으로 구동되는 구조 예측의 혁신은 단백질 과학에서 이러한 모델의 힘을 입증했지만 충분한 열역학 데이터를 수집하는 것이 항상 주요 장애물이었습니다. cDNA 디스플레이 단백질 분해의 규모와 효율성으로 인해 수백만 개의 작은 도메인에 대한 안정성을 측정하는 데 있어 주요 한계는 DNA 합성 및 시퀀싱 비용입니다. 이 두 가지 모두 빠르게 감소하고 있습니다. 단백질 서열 공간의 크기와 다양성은 생물학 및 단백질 설계에 엄청난 도전을 야기합니다. cDNA 디스플레이 단백질 분해는 이 공간에서 폴딩 안정성의 대규모 매핑을 위한 강력한 접근 방식을 제공합니다.