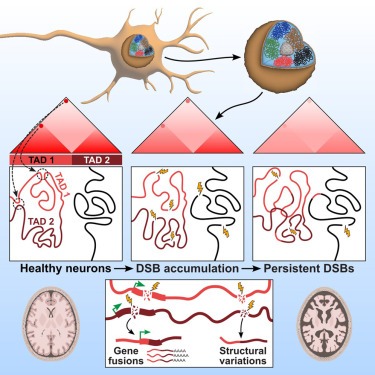
Neuronal DNA double-strand breaks neurodegeneration에서 genome structural variations과 3D genome disruption로 이어집니다.
Abstract
neurons의 Persistent DNA double-strand breaks (DSBs)는 알츠하이머병(AD)을 포함한 neurodegenerative diseases의 early pathological hallmark로, genome integrity를 방해할 수 있습니다.
우리는 human postmortem prefrontal cortex samples에서 single-nucleus RNA-seq을 사용하여 AD의 excitatory neurons이 somatic mosaic gene fusions이 풍부하다는 것을 발견했습니다. Gene fusions은 특히 DNA damage repair 및 senescence gene signatures를 가진 excitatory neurons에서 강화되었습니다. 또한, neurodegeneration의 CK-p25 마우스 모델에서 DSB로 burdened neurons에서 somatic genome structural variations과 gene fusions이 강화되었습니다. DSB가 풍부한 neurons은 또한 synaptic, neuronal development, and histone genes의 transcriptional changes와 일치하는 3D genome organization의 progressive multiscale disruption과 함께 cohesion level이 높았습니다.
전반적으로 이 연구는 neurodegenerative diseases 진행의 pathological steps으로서 neurons에서 DSB에 의한 genome stability와 3D genome organization의 disruption을 보여줍니다.
Figure
DSB-bearing neurons은 neurodegeneration에서 mosaic genome structural variations을 가지고 있습니다.
- γH2AX가 enriched된 neurons이 genome structural variation 증가의 대리인으로서 gene fusions levels이 증가했는지 테스트
- gene fusions의 증가가 genome-wide genome structural variations가 전반적으로 증가한 결과인지 확인
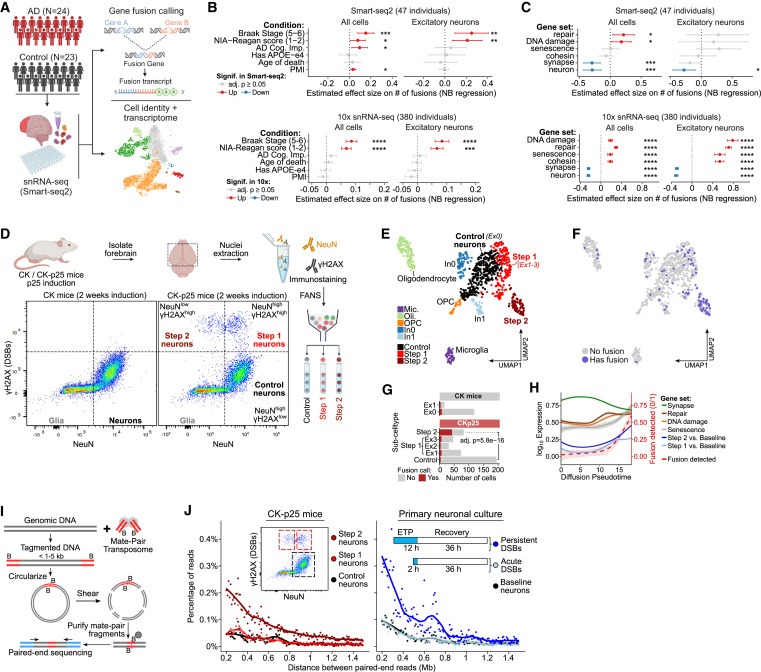
Figure 1. neurons의 DSB는 mosaic genome structural variations으로 이어집니다.
(A) 인간 사후 뇌에서 snRNA-seq(Smart-seq2) 분석의 모식도.
(B) human sample covariates의 function으로서 human snRNA-seq을 사용한 number of gene fusions의 effect size.
(C) (B)와 유사한, different Gene Ontology (GO) terms과 연관된 gene sets의 function으로서의 number of gene fusions의 effect size.
(D) NeuN (neuron) 및 γH2AX(DSB) marker를 사용하여 2주간 유도한 후 CK/CK-p25 forebrain의 대표적인 FANS dot-plot.
(E) induction 1주 또는 2주 후 CK/CK-p25 snRNA-seq의 UMAP embedding.
(F) 최소 1개 이상의 gene fusion event (파란색)를 포함하는 cells을 snRNA-seq UMAP embedding에 오버레이한 모습.
(G) 하나 이상의 gene fusion이 있는 number of cells과 Control, Step 1 (Ex1, Ex2, Ex3 clusters), and Step 2에 대한 total number of cells의 정량화.
(H) Control, Step 1, and Step 2의 Trajectory analysis.
(I) mate-pair sequencing의 개략도.
(J) genome structural variation events를 나타내는 200kb 이상의 large genomic distances로 분리된 read pairs의 비율을 정량화한 mate-pair sequencing quantifying Plots.
[Figure 1A] 47명 전체에 대해 well-based single-nucleus RNA-seq full-transcript profiles (snRNA-seq)을 사용하여 6,180개의 세포를 얻음.
[Figure 1B] AD pathology individuals의 세포에서 gene fusions이 non-AD individuals의 세포에 비해 유의미하게 enriched되어있는 것을 발견. 또한 prefrontal cortex에 neurofibrillary tangles (NFT)의 존재 여부에 따라 late-AD individuals의 세포에서 gene fusions이 유의하게 enriched되어있는 것을 발견함.
[Figure 1B] cell type별로 gene fusions enrichment를 Stratifying하면, 특정 excitatory neurons이 gene fusions에 대해 enrichment되어 있으며, late AD individuals에서 다른 모든 cell type에 비해 더 큰 effect size를 보이는 것으로 나타남.
[Figure 1C] DNA damage, repair, and senescence-associated gene sets의 발현이 높은 excitatory neurons은 10x datasets에서 gene fusion burden 크게 증가한 것으로 나타남.
반면, neuronal identity 및 synaptic function과 관련된 gene의 발현이 높은 excitatory neurons은 두 데이터 세트 모두에서 gene fusion에 대한 burden이 감소.
[Figure 1C] 모든 세포를 함께 분석한 결과, repair, DNA damage, and senescence-associated cells에서 fusions이 enrichment된 경우 same directions으로 유의미한 효과가 나타났지만 effect sizes (in the 10x data)는 감소한 것으로 나타남.
- 이러한 expression profiles을 가진 세포에서 mosaic gene fusion burden이 증가한다는 것은 neurons의 DNA breaks이 genome structural variations이 증가하여 senescence-like state로 이어질 수 있음을 시사.
[Figure 1D] CK-p25 마우스에서 2주 동안 p25를 induction 후, (1) high levels의 NeuN과 baseline levels의 γH2AX를 가진 neurons(Control neurons), (2) high levels의 NeuN과 γH2AX를 가진 neurons(Step 1 neurons), (3) low levels의 NeuN과 high levels의 γH2AX를 가진 세포(Step 2 neurons)를 분리.
- 이전에 Step 2 neurons이 실제로 senescence and immune genes의 발현을 특징으로 하는 neurodegeneration의 later step에 진입하는 neurons이라는 것을 보여줌.
[Figure 1E] CK-p25 마우스의 Step 2 neurons은 뚜렷한 expression cluster를 형성한 반면, Step 1 neurons은 UMAP에서 Control (Ex0)과 2 Step 2 clusters 사이에 three clusters (Ex1, Ex2, Ex3)로 분포되어 있음.
[Figure 1F,G] Step 2 neurons은 gene fusion calls에 대해 유의미하게 enriched해진 반면, Step 1 neurons은 gene fusion이 소폭 증가한 것으로 나타남.
[Figure 1H] gene fusion frequency는 Step 2 signature와 유사하게 senescence와 관련된 genes과 함께 pseudo time의 함수로 증가. 반면에 DNA damage and repair와 관련된 genes은 Step 1 signature와 유사하게 증가 및 정체
- 이러한 결과는 DSB burden이 증가한 neurons (Step 1)이 gene fusions 증가를 특징으로 하는 노 senescence-like state (Step 2)로 진행됨을 보여줌.
[Figure 1I] genomic DNA는 biotinylated adaptors를 운반하는 Tn5 transposase에 의해 fragmented 시킨 후, fragments를 circularized한 다음 200-300bp fragments으로 절단. 그런 다음 biotin pull-down and paired-end sequenced를 통해 ligation junctions을 enriched함.
[Figure 1J] 놀랍게도, Step 1 and Step 2로 분할된 γH2AXhigh neurons의 mate-pair sequencing 결과, Step 2 neurons의 structural variation events frequency가 훨씬 높은 반면(241% 증가), Step 1 neurons의 structural variation frequency는 Control neurons과 유사.
- 이러한 결과는 Step 1 neurons에서 persistent DSB와 delayed repair가 repair errors의 가능성을 높이고 genome structural variations이 증가한 Step 2 neurons으로 전환될 수 있음을 시사.
- 이러한 결과를 종합하면 excitatory neurons에서 persistent DSB로 인한 delayed repair가 mosaic genome structural variation의 accumulation으로 이어진다는 것을 알 수 있음.
Highly expressed genes과 long genes은 genome structural variations에 취약한 부위입니다.
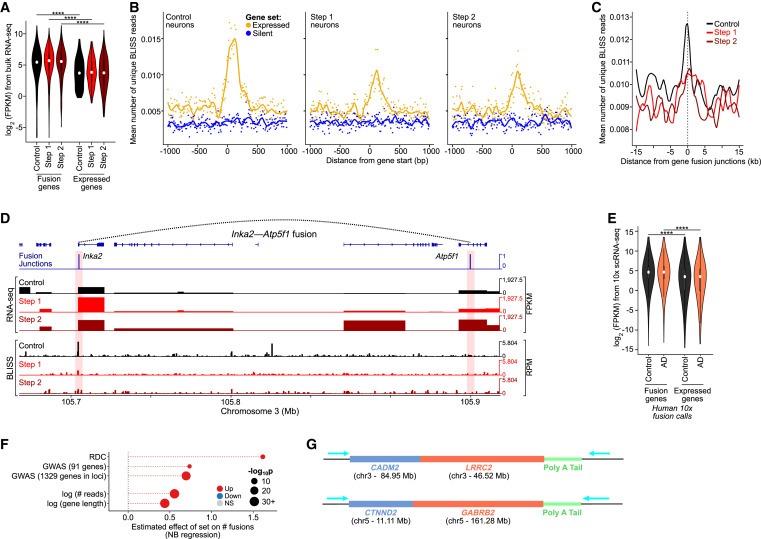
Figure 2. Highly expressed genes과 long genes은 neuron의 genome structural variations에 취약한 부위입니다.
(A) CK-p25 gene fusions에 관여하는 genes의 Expression level (Benjamini–Hochberg correction을 통한 Wilcoxon test, ∗∗∗∗p < 1e-4).
(B) Control, Step 1, and Step 2 neurons에 대한 상위 2,500개의 expressed genes(노란색)와 하위 2,500개의 silent genes (파란색)에 대한 gene starts 지점을 중심으로 한 BLISS reads의 Aggregate plots.
(C) CK-p25 neurons에서 snRNA-seq에 의해 호출되는 gene fusion junctions을 중심으로 한 BLISS reads의 Aggregate plots.
(D) CK-p25 mice Step 2 neurons에서 example gene fusion의 Genome browser snapshot.
(E) human gene fusions에 관여하는 genes의 Expression level (10x snRNA-seq) 대 all expressed genes (Wilcoxon test with BH correction).
(F) RDC gene, AD GWAS genes, gene expression level (reads), and gene length (bp)로 식별된 genes의 function에 따른 number of gene fusions의 effect size.
(G) ONT long-read cDNA sequencing을 통해 확인된 example gene fusions의 모식도.
[Figure 2A] Control, Step 1, and Step 2 neurons의 fusion genes이 neurons에서 발현된 genes보다 훨씬 더 높은 발현을 보임.
- 이는 transcription 증가가 genome structural variation 증가와 관련이 있음을 시사
[Figure 2B] Control, Step 1, and Step 2 neurons에서 Break Labeling in situ and sequencing (BLISS)을 사용하여 DSB를 직접 매핑한 결과, 높은 gene expression이 Control, Step 1, and Step 2 neurons에서 DSB 증가와 밀접한 관련이 있음을 발견.
highly transcribed genes의 DSB enrichment level은 Control에 비해 Step 1, and Step 2 neurons에서 더 낮음.
- 이는 highly transcribed genes이 DSB의 hotspots인 반면, Step 1에서 증가한 DSB는 genome 전체에 확률적으로 분포되어 있음을 시사.
[Figure 2C] Control cells에서 highest enrichment를 보이는 gene fusion junctions에서 DSB가 enrichment되는 것을 관찰했는데, 이는 gene-fusion junctions이 DSB가 빈번하게 발생하는 위치임을 나타냄.
[Figure 2D] fusion junction-spanning fragments 수에 따른 top gene fusion calls의 예(Inka2-Atp5f1 및 Cdip1-Ubald1)는 fusion junctions에서 gene expression과 DNA breakpoints가 증가한 것으로 나타남.
[Figure 2E] CK-p25의 BLISS에서 관찰한 것과 유사하게, excitatory neurons의 human fusion genes도 AD와 control 모두에서 gene expression이 증가하는 경향이 있음.
[Figure 2F] fusions에 관여하는 genes과, synaptic function과 cell-cell adhesion에 중요하고 neural precursor cells에 recurrent DSB clusters (RDC)를 보유하는 것으로 알려진 actively transcribed되는 long neural genes set 사이에 상당한 중복이 있음을 확인.
이러한 RDC genes의 length와 active transcription은 DSB 및 gene fusion에 대한 propensity 증가를 주도할 가능성이 높음. 모든 genes에서 높은 transcription levels과 gene length 증가는 모두 gene fusions 증가와 유의미한 연관성이 있었음.
[Figure 2G] RDC genes (예: CADM2, CTNND2)를 포함한 여러 gene fusions을 확인.
- 이러한 결과를 종합하면, long genes와 highly transcribed genes와 관련된 DNA breaks이 증가하면 structural variation과 gene fusion에 취약해진다는 것을 알 수 있음.
DSB burdened Neurons은 3D genome organization의 disruption과 관련이 있습니다.
- DSB를 가진 excitatory neurons에서 cohesin complex의 levels을 조사
- DSB와 chromatin structural proteins의 변화가 3D genome organization에 영향을 미칠 수 있는지 조사
- DSB와 cohesin의 misregulation이 loop와 TAD organization에 어떤 영향을 미치는지 확인.
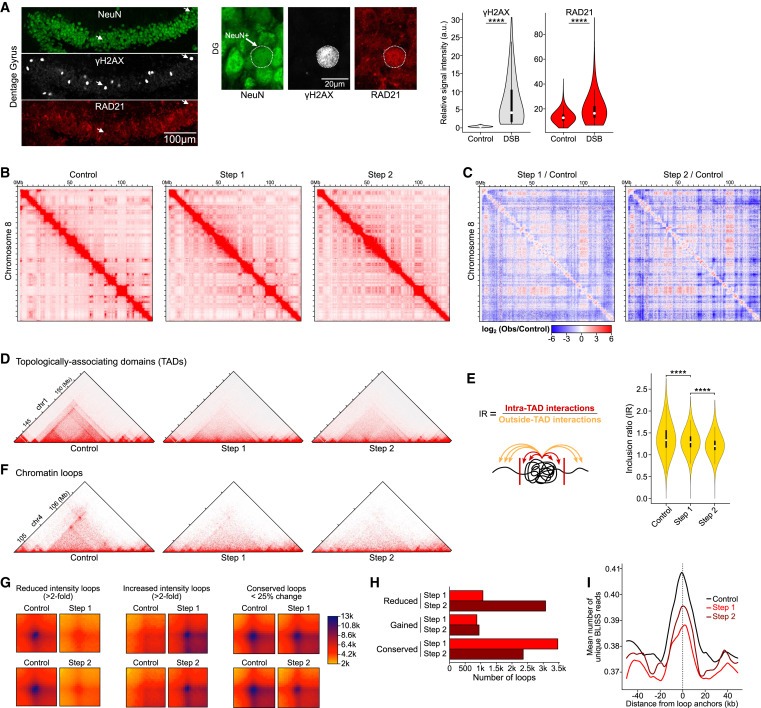
Figure 3. DSB burdened Neurons은 multiple scales에서 3D genome organization이 global disruption되는 현상을 보입니다.
(A) 2-week-induced CK-p25 mice의 baseline DSB를 가진 neurons과 DSB enriched된 neurons에 대한 representative immunohistochemistry images 및 RAD21 (cohesin subunit)의 정량화.
(B) Full chromosome (chr8) Hi-C chromatin interaction heatmaps.
(C) Step 1/Control and Step 2/Control을 비교한 Differential heatmaps.
(D) TAD의 disruption을 보여주는 Representative Hi-C interaction plots.
(E) inclusion ratio (IR)을 통한 TAD disruption의 정량화.
(F) chromatin loops의 disruption을 보여주는 Representative Hi-C interaction plots.
(G) Step 1 vs. Control과 Step 2 vs. Control의 chromatin loops 감소, 증가 및 conserved에 대한 Aggregate heatmaps.
(H) Step 1 (빨간색) 및 Step 2 (어두운 빨간색)에서 chromatin loop disruption의 정량화를 나타내는 Bar plot.
(I) CK-p25 neurons의 loop anchors를 중심으로 한 BLISS reads의 Aggregate plots.
[Figure 3A] DSB가 있는 neurons은 baseline DSB가 있는 neurons에 비해 RAD21이 significantly enriched됨.
[Figure 3B,C] Hi-C는 long-range interactions의 증가와 감소를 모두 포함하는 3D genome organization에서 genome-wide alterations을 밝혀냄. Step 1 neurons에서 long-range interactions의 intensity가 1Mb 이상으로 net decrease함. 이는 Step 2 neurons에서 더욱 두드러져 long-range interactions의 intensity가 0.5Mb부터 감소함.
[Figure 3D,E] Step 1에서 TAD definitions이 전반적으로 크게 감소했으며 Step 2에서 더 악화됨.
[Figure 3F] control neurons과 비교하여 Step 1 and Step 2 neurons에서 chromatin loops가 파괴되는 것을 관찰.
[Figure 3G] loops의 Aggregate plots은 loop intensity의 감소와 증가를 모두 포함하는 genome-wide disruption of loops를 보여줌
[Figure 3H] 더 많은 loops가 intensity의 증가보다 감소를 보였으며, loop loss는 Step 1 neurons보다 Step 2 neurons에서 훨씬 더 두드러짐.
[Figure 3I] 우리는 Control, Step 1, and Step 2의 loop boundaries에서 DSB가 enrichment되는 것을 발견. 이는 loop boundaries가 T세포와 B세포에서 TOP2 B-mediated DNA breaks의 hotspots이라는 관찰과 잘 일치.
[Figure 3I] loop boundaries에서 DSB의 enrichment는 Step 1에서 가장 낮았고 Step 2가 그 뒤를 이었는데, 이는 Step 1에서 DSB burden이 확률적으로 증가하고 Step 2에서 부분적으로 복구되는 것과 일치.
neurons에서 DSB를 유도하면 3D genome organization을 disrupt하기에 충분합니다.
- etoposide로 DSB를 유도하면 3D 게놈 조직에도 변화가 생기는지 확인.
- etoposide-induced DSBs가 cohesion levels에 미치는 영향을 측정
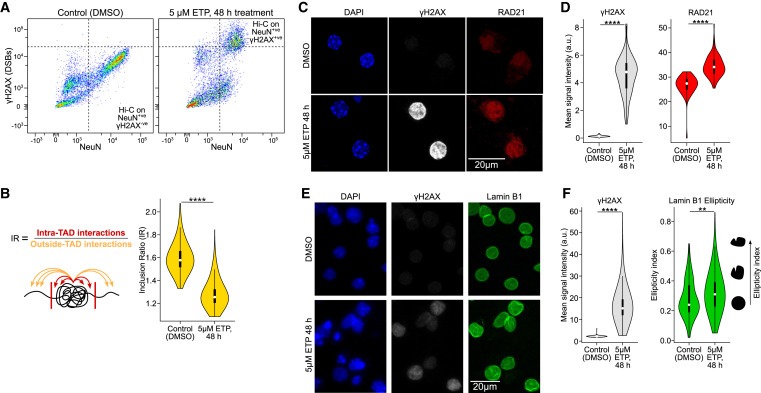
Figure 4. neurons의 DSB는 3D genome organization을 disrupt하기에 충분합니다.
(A) primary neuronal culture에서 etoposide (48시간 동안 5μM) 또는 vehicle (DMSO) 처리 후 NeuN 대 ϒH2AX immunoreactivity의 FANS dot-plots.
(B) TAD disruption의 정량화 (IR, Wilcoxon test, ∗∗∗∗p < 1e-4).
(C) etoposide (48시간 동안 5μM) 또는 vehicle treatment (DMSO) 후 primary culture neurons (DIV 12)에서의 RAD21(cohesin subunit) IHC의 Representative images 및 정량화.
(D) control neurons과 etoposide-treated neurons의 ϒH2AX levels (회색) 및 RAD21 levels (빨간색)에 대한 이미지를 정량화한 Violin plots (Wilcoxon test).
(E) etoposide (48시간 동안 5μM) 또는 vehicle treatment (DMSO) 후 primary culture neurons (DIV 12)에서의 Lamin B1 IHC의 Representative images.
(F) control neurons과 etoposide-treated neurons의 ϒH2AX levels (빨간색) 및 lamin B1 ellipticity (녹색)에 대한 이미지를 정량화한 Violin plots (Wilcoxon test, ∗∗p < 0.01, ∗∗∗∗p < 1e-4).
[Figure 4A] etoposide treatment 군에서 high γH2AX를 가진 NeuN+ve neurons과, DMSO 처리에서 baseline γH2AX를 가진 NeuN+ve neurons을 FANS로 분류.
[Figure 4B] inclusion ratio를 사용하여 TAD integrity를 정량화한 결과, etoposide-treated primary neuronal cultures에서 DSB burden이 높은 CK-p25 마우스의 in vivo neurons과 유사하게 유의미한 감소가 나타남.
[Figure 4C,D] DSB burden이 높은 CK-p25 neurons과 마찬가지로, etoposide-mediated DSBs를 보유한 neurons에서 RAD21 수치가 유의하게 상승.
[Figure 4E,F] etoposide로 처리한 primary neurons에서 Lamin B1을 염색한 결과, CK-p25 마우스 모델과 유사하게 nuclear envelope에서 Lamin B1이 파괴된 것을 관찰.
- 이러한 결과를 종합하면 neurons의 DSB가 3D genome organization을 방해하기에 충분하다는 것을 알 수 있음.
DSB에 의한 3D genome organization의 disruption은 differential gene regulation과 관련이 있습니다.
- Step 1 and Step 2 neurons의 chromatin loop dysregulation이 gene expression changes와 관련이 있는지 조사
Figure 5. DSB-associated dysregulation of the 3D genome은 differential gene expression과 일치합니다.
(A) DSB가 있는 neurons에서 differential loops와 differential gene expression을 비교한 모식도.
(B) Step 1과 Step 2 모두에서 loop loss에 대한 loop fold change 대비 상승(녹색) 및 하락(빨간색) DEG의 백분율.
(C) loop loss-associated down-regulated genes의 Gene ontology analysis (GORILLA).
[Figure 5A] fold-change thresholds가 증가함에 따라 differential loops를 stratified하고 Control, Step 1, and Step 2 neurons의 bulk RNA-seq을 사용하여 이러한 loops 근처 (anchors로부터 +/- 5 kb)에서 differential up and down-regulated genes의 비율을 계산.
[Figure 5B] loop intensity의 감소는 Step 1 and Step 2 neurons 모두에서 더 높은 비율의 down-regulated genes과 더 낮은 비율의 up-regulated genes와 밀접한 관련이 있음.
[Figure 5C] loop loss와 관련된 Down-regulated genes은 synaptic, cell adhesion, and neuronal development genes에서 유의미하게 enriched되어있는 반면, intensity가 증가한 loops는 differential gene expression과 일관된 관계가 없었음.
- 이는 loop gain과 gene expression 사이의 연관성이 상황에 따라 다르다는 것을 시사
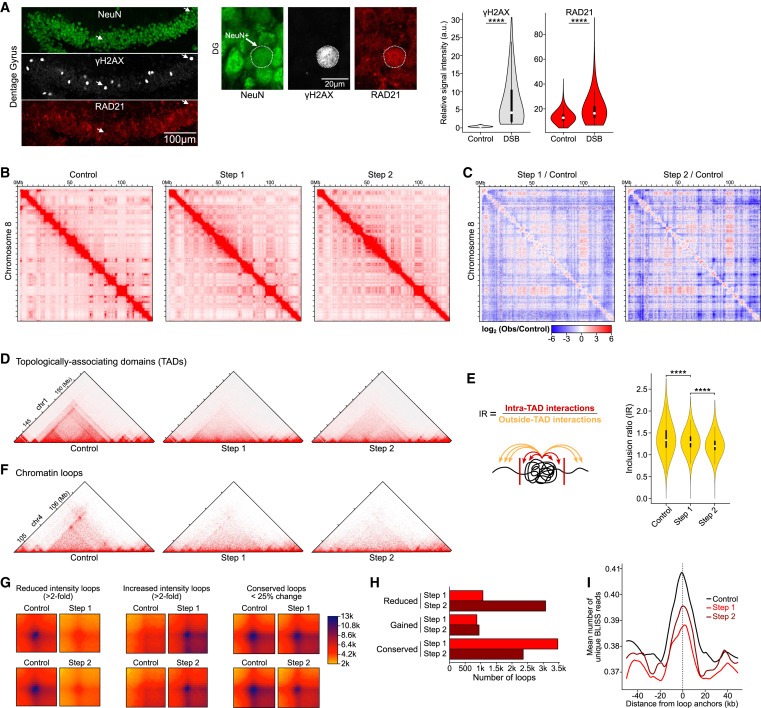
Figure 6. 알츠하이머병에서 cohesin 및 histone genes의 Up-regulation.
(A) signature gene sets에 대한 Control, Step 1, and Step 2의 bulk RNA-seq에서 Z scores로 표시된 differential gene expression의 Heatmap.
(B) Mathys et al. (∗differential) 의 10x snRNA-seq에서 excitatory subtypes에 걸쳐 histone 및 cohesin genes의 differential gene expression Heatmap
(C) 알츠하이머병과 Control human postmortem tissue에서 RAD21의 Representative immunostaining images.
(D) RAD21 staining은 이미지당 높은 RAD21을 가진 neurons의 백분율로 정량화하거나(왼쪽), RAD21 mean intensity (오른쪽)를 사용하여 정량화.
(E) histone gene cluster (녹색 블록, chr13)에 대한 Hi-C interaction matrix.
(F) Step 1/Control and Step 2/Control을 비교한 Differential Hi-C heatmaps.
[Figure 6A] Step 1 and Step 2 neurons의 differential genes 중 cohesin complex genes이 이전에 보고된 여러 DNA damage, senescence, and immune response genes과 함께 up-regulated되는 것을 발견.
[Figure 6B] Cohesin complex genes은 427명에 걸친 10x snRNA-seq data에서 AD pathology가 높은 피험자의 excitatory neurons에서 consistently and significantly up-regulated됨.
[Figure 6C,D] immunohistochemistry를 사용하여 human postmortem tissue에서 cohesin subunit RAD21의 levels을 측정한 결과, AD pathology를 가진 피험자의 neurons에서 RAD21의 levels이 크게 증가함.
[Figure 6E,F] histone gene의 gene expression이 증가함에 따라 13번 chromosome의 histone gene clusters는 Step 1 and Step 2 neurons에서 de novo loops를 얻음
- histone gene clusters의 upregulation의 근간이 되는 3D genome level regulation을 강조.
- 이러한 결과를 종합하면 DSB-mediated 3D genome changes와 neurodegeneration의 근간이 되는 gene regulation program 간의 연관성이 입증됨.
Disscussion
neurodegeneration의 CK-p25 모델에서 neurons은 DSB가 증가하는 initial step (Step 1)를 거쳐 gene fusions과 structural variation이 enriched해지고 senescence genes으로 표시되는 later step (Step 2)로 진행됩니다. 우리는 neurons의 persistent DSB가 etoposide에 의한 DSB induction과 일치하는 genome structural variations을 유발한다는 가설을 세우고, persistent but not acute DSB induction은 structural variations을 증가시키지 않는다고 가정하고 있습니다. non-neuronal cells and neural precursor cells에서 관찰된 이전 결과와 일치하는 Gene fusion junctions은 DSB에 대해 enriched하게 나타났고, genes with higher expression and length에 대해 fusions이 enriched하게 나타났습니다. 놀랍게도 Step 1에서는 highly transcribed and long genes에 대해 lower DSB enrichments를 보이며, DSB는 더 적은 UMI로 지원되어 Step 1 DSB가 대부분 확률적임을 시사합니다. 대조적으로, Step 2의 UMI는 controls과 유사하게 분포되어 있어 structural variations의 대가로 DSB의 repair가 증가했음을 시사합니다. 우리는 증가된 확률론적 DSB burden이 DNA repair resources에 burden을 주어, highly expressed genes, long genes, and loop anchors와 같은 DNA breaks의 hotspots에 불균형적으로 영향을 미치는 defective DNA repair and genome structural variations을 초래할 수 있다고 제안합니다. postmortem tissue에서 직접 DSB를 매핑하는 향후 연구에서는 알츠하이머병의 structural variations과 관련된 추가 hotspots을 밝혀낼 수 있습니다.
linear genome의 disruptions에 더하여, 우리는 neurons에서 DSB-mediated senescence-like state가 neurodegeneration에서 3D genome의 혼란으로 이어진다는 것을 보여줍니다. 우리는 특히 Step 2 senescence-like neurons에서 neuronal identity와 관련된 여러 genes의 differential regulation과 함께 TAD definition 및 loop intensities의 general loss를 관찰했습니다. 이는 특히 excitatory neurons과 oligodendrocytes에서 나타나는 epigenome erosion과 놀라울 정도로 유사하며, companion manuscript에서 보고된 cell-type-specific chromatin accessibility profile의 loss가 특징입니다. 흥미롭게도 최근 brain aging and cognitive decline를 동반한 inducible DNA damage mouse model (ICE mouse model)에서 epigenetic information, TAD insulation 및 cell identity의 erosion이 보고되었습니다. 최근 연구에 따르면 DSB가 loop extrusion의 장애물 역할을 할 수 있다고 제안했습니다. 인접한 DSB가 양쪽에서 premature loop extrusion block을 유발하여 chromatin loops와 더 큰 도메인을 방해할 수 있다고 생각할 수 있습니다. 놀랍게도 X-ray radiation-induced DSBs는 fibroblasts에서 TAD의 segregation을 증가시켰습니다. 향후 연구에서는 irradiation에 의해 형성된 acute DSB와 persistent DSB가 genome organization에 어떤 영향을 미치는지, 또한 post-mitotic cells과 dividing cell이 DSB에 대한 반응이 어떻게 다른지 탐구해야 합니다. 또한 DSB가 enriched 된 neurons에서 nuclear structural proteins, cohesin, and Lamin B1 levels에서 DSB-mediated changes를 관찰했는데, 이는 이전에 AD neurons과 senescent astrocytes에서 관찰한 것과 유사합니다. Lamin B1의 disruption에도 불구하고 genome의 A / B compartmentalization은 상대적으로 영향을 받지 않았으며, 이는 망막 막대 뉴런에서 관찰 된 것과 일치하며, nuclear lamina와의 heterochromatin interactions의 loss에도 불구하고 phase separation이 A / B compartments formation을 유도합니다.
3D genome의 변화는 전반적으로 transcriptional changes와 일치했으며, loop intensities의 감소는 genes의 down-regulation과 현저한 상관관계가 있었습니다. 흥미롭게도, 여러 histone gene의 upregulation과 일치하는 histone gene clusters에서 loops가 새롭게 형성되는 것을 관찰했습니다. 이전 연구에서는 DNA damage response, senescence, brain aging, neuronal identity, and memory formation에서 histones의 중요성이 밝혀졌습니다. 이러한 결과는 neurons의 3D genome organization과 neurodegenerative state와 관련된 transcriptional programs 사이의 연관성을 강조합니다. 실제로 chromatin loops의 증가는 histone gene up-regulation과 관련이 있지만, 다른 independent mechanisms of gene regulation이 degenerating neurons에서 genes up-regulation의 근간이 될 수 있습니다. 또한, dividing cell types에서 senescence-associated 3D genome reorganization이 나타난 연구도 있습니다. 우리의 연구 결과는 senescence-like state의 neurons에서 3D genome organization changes가 neurodegeneration에서 genome dysfunction을 매개할 수 있다는 것을 보여줍니다.
전반적으로, 우리의 연구는 DNA double-strand breaks이 mosaic genome structural variations과 neurons의 3D genome organization로 이어진다는 것을 보여줍니다. 우리는 알츠하이머병에서 cohesin, DNA damage, and senescence-like gene expression과 관련된 excitatory neurons의 genome structural variations으로 인한 mosaic gene fusions 증가를 관찰합니다. 이러한 데이터는 neuronal genomic integrity과 3D genome organization을 neurodegeneration의 근원이 되는 cellular pathologies와 연결합니다.
Limitations of the study
Gene fusions은 genome structural variations의 subset에서 발생하며, neurodegeneration에서 DNA damage-dependent genome structural variations의 전체 범위를 해결하려면 single-cell RNA sequencing과 long-read single-cell genome sequencing을 결합하는 미래 기술이 필요할 것입니다. Single-cell RNA-seq는 gene expression에 대한 sparse readout을 제공하며 일반적으로 각 세포의 gene fusions 수를 과소평가할 가능성이 높습니다. 비교적 드물기는 하지만, library preparation 중 PCR artifacts와 random ligations을 통해 발생하는 chimeric molecules은 진정한 mosaic gene fusions에서 발생하는 chimeric molecules과 구별하기 어렵습니다. 우리는 여기서 살펴본 AD 대 Control classification과 다양한 gene expression pattern enrichments가 library preparation 중 PCR artifacts와 random ligations을 수정하지 않는다고 가정합니다. 3D genome organization의 disruption은 해결되지 않은 DSB와 genome structural variations에 의해 직접적으로 발생할 수 있습니다. 현재 연구에서는 이러한 각 구성 요소의 정확한 역할을 확인할 수 없습니다. unresolved DSBs locations과 post-repair locations에서 targeted Hi-C를 수행하는 향후 연구를 통해 이러한 질문에 더 잘 답할 수 있을 것입니다.