
확장된 1000 Genomes project: 602개의 trio를 대상으로 High-coverage whole-genome sequencing 적용

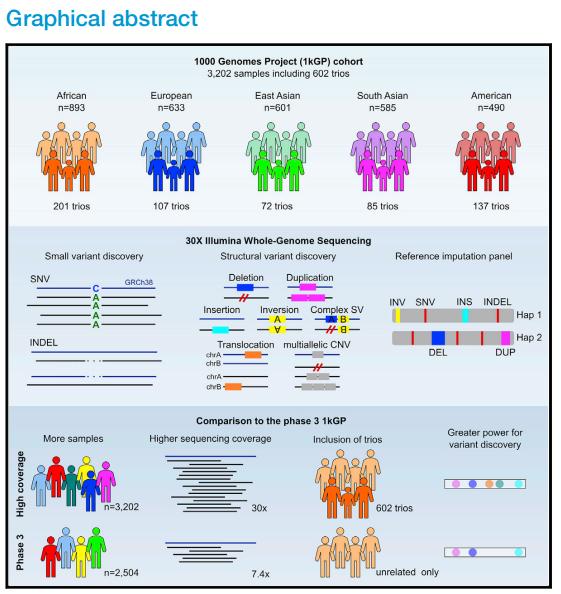
1000 Genome Project(1kGP)는 공공에 완전 개방된 전장 유전체 해독(Whole Genome Sequencing) 자료 리소스 중 가장 큰 리소스입니다. 최종 배포본인 phase 3 release(1kGP)는 26개의 인구에서 서로 관련이 없는, 즉 후술할 trio 단위의 연구와 다르게 개개인 간 유전적 관련성이 없는 샘플 2,504개를 포함했으며, 주로 low-coverage WGS를 기반 삼았습니다. 이에 해당 연구는 Illumina를 사용하여 30X depth로 sequencing된 602개의 완전한 trio를 포함하는, high-coverage 샘플 3,202개를 포함하는 WGS 1kGP 리소스를 소개합니다. 주로 single-nucleotide variant(SNV), short insertion and deletion(INDEL)을 발견했으며, 머신 러닝 모델을 통해 여러 가지의 분석 방법을 통합하여 포괄적인 structural variants(SV)를 생성하였습니다. 이 연구를 통해 나온 확장된 1000 Genome Project 데이터는 phase 3에 비해 variant calls의 sensitivity와 precision가 향상되었음을 보여주며, 특히 넓은 frequency spectrum에서 rare SNVs 및 INDELS, 그리고 SV의 variant call sensitivity & precision이 향상되었음을 확인했습니다. 또한, 보다 발전된 reference imputation panel을 생성하여, 여기서 발견된 variants가 연관성 연구에 사용될 수 있을 것을 기대합니다.

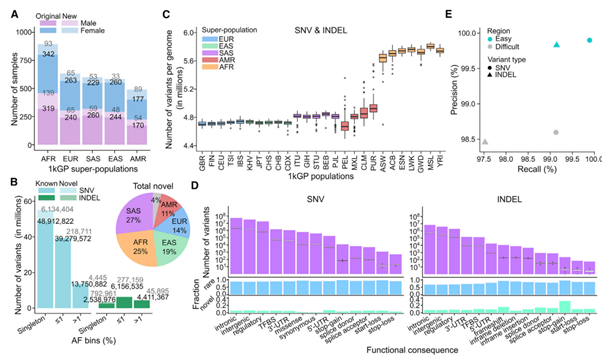
[Figure 1] 3,202 1kGP sample high-coverage WGS data에서의 SNV/INDEL 발견
(A) 성별 및 super-population에 따라 나누어 분류한 샘플 수입니다.
기존: 2,504 1kGP samples / 신규: 698개의 sample 추가
(B) 3,202개의 샘플에서 발견한 코호트 단계 alternative allele counts. AF bins(Allele frequency 구간)로 나누었습니다.
Novel / known: dbSNP build 155(해당 논문 이전의 1000 Genome Project 버전)에 존재하지 않는/존재하는 sites입니다.
AF는 서로 연관되어 있지 않은 2,504개의 샘플을 기준으로 추정했습니다.
Pie chart: super-population ancestry에 따른 novel variants의 구분. 회색 부분은 해당 샘플이 하나 이상의 super-population에 속함을 의미합니다.
(C) 각 genome 별로 small variant loci의 개수를 세었습니다. 인구를 기준으로 나누었습니다.
(D) 예측된 functional SNVs, INDELs(상염색체 내)입니다.
위: 코호트 단계에서 variants의 수를 센 막대그래프, 그리고 샘플 단계에서 그 분포를 box plot으로 함께 나타낸 그림입니다. 2,504개의 서로 관련 없는 샘플을 대상으로 진행했습니다.
중간: 예측된 functional sites 중 rare(MAF(Minor Allele Frequency) ≤ 1%) SNVs & INDELs의 비율을 나타냅니다. 해당 논문에서 강조하는 부분으로, 예측된 functional SNV 중 86%~97%, INDEL 중 67% ~ 95%가 rare genetic variants로 분류되었습니다. 이 중 SNV는 1~7%가, INDEL은 5~32%가 novel하게 분류되었습니다.
(E) GIAB(Genome in a Bottle: Consortium) truth set v3.3.2에 의존하여 계산된 Precision vs Recall. Genome의 easy / difficult한 region 으로 나누었습니다.
Easy & difficult region은 GIAB에 의해 정의되었으며, difficult region이 전체 genome의 20%에만 해당하지만, 높은 비율의 multiallelic states를 포함하여 변이의 주요 사이트라고 볼 수 있습니다. 뿐만 아니라, INDEL loci도 다수 포함하고 있어 특정 allele이 변이되었을 뿐만 아니라 추가 혹은 제거되어 있을 확률이 높습니다.
(Super-population ancestry 표기: EUR(유럽인), AFR(아프리카인), EAS(동아시아인), SAS(서아시아인), AMR(아메리카인))

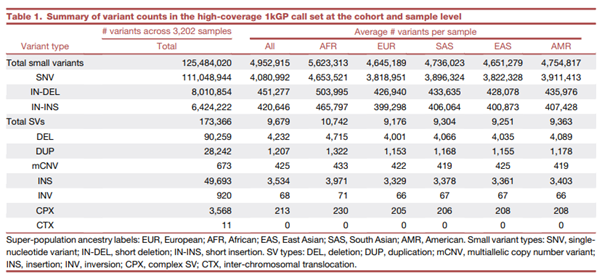
[Table 1] 코호트 & 샘플 단계 각각 high-coverage 1kGP call set의 variant count에 대한 요약 테이블
(용어) Super-population ancestry 표기: EUR(유럽인); AFR(아프리카인); EAS(동아시아인); SAS(서아시아인); AMR(아메리카인). Small variant 종류 표기: SNV(Single Nucleotide Variant); IN-DEL(short deletion); IN-INS(short insertion); Structural Variant 종류 표기: DEL(deletion); DUP(duplication); mCNV(multiallelic CNV); INS(insertion); INV(inversion); CPX(complex SV); CTX(inter-chromosomal translocation)

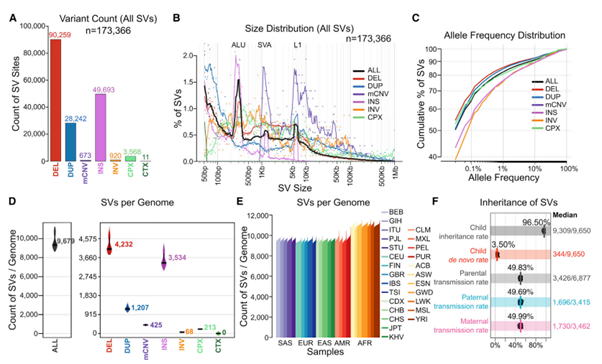
[Figure 2] 3,203 1kGP 샘플의 high-coverage WGS data에서의 SV 발견
(A-C) 각 SV class의 (A) 수, (B) 크기에 대한 분포, (C) 대립 유전자 빈도(allele frequency) 분포표
(D-F) SV의 sample count 당 평균. (D) variant class, (E) ancestral population, (F) 유전을 주고받는 비율.
(F)에서, child inheritance rate는 아이가 가지고 있는 SV 중 부모에게 물려 받은 비율을 의미합니다. Parental transmission rate는 부모의 genome에서 아이에게 전달되는 비율을 의미합니다. 해당 figure에 표현된 부분은 모두 informative SV이며, 한 명의 부모에게서만 이형(heterozygous)인 조건을 가지고 있습니다. 세로로 표시된 줄은 각각 평균 값을 의미하고, 오른쪽에는 median SV counts를 표현합니다.
(용어) Super-population ancestry 표기: EUR(유럽인); AFR(아프리카인); EAS(동아시아인); SAS(서아시아인); AMR(아메리카인); Structural Variant 종류 표기: DEL(deletion); DUP(duplication); mCNV(multiallelic CNV); INS(insertion); INV(inversion); CPX(complex SV); CTX(inter-chromosomal translocation)

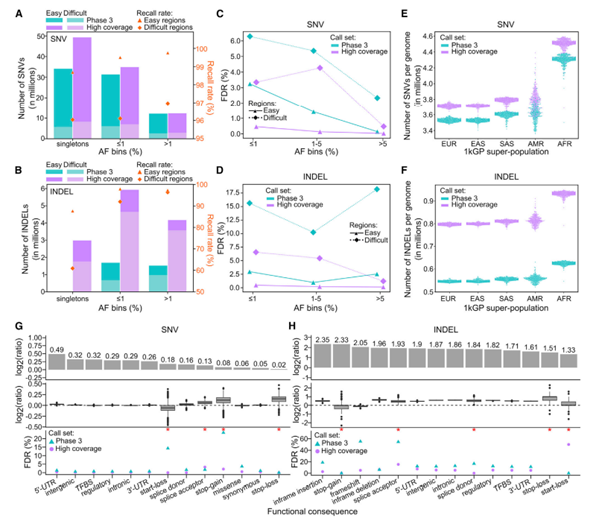
[Figure 3] small variant call과 phase 3 call set 간의 비교
(A, B) phase 3의 2,504개의 샘플 및 high-coverage dataset에서 보유하는 SNV 수 (B) INDEL 수의 요약입니다. AF bins, genome region에 의해 나누었습니다.
(C, D) FDR(False Discovery Rate)의 비교 ((C) SNV, (D) INDEL) 입니다. AF bins, genome region에 의해 나누었습니다. GIAB truth set에 의해 정의된 easy & difficult region 모두 새로운 버전의 1000 Genome Project에서 FDR이 낮아, 실제로 SNV이나 INDEL이 아닌데도 그렇게 분류하는 경우가 줄어든 것을 확인할 수 있습니다.
(E, F) Sample-level (E) SNV, (F) INDEL 수입니다. 1kGP super-population ancestry에 의해 나누었습니다.
(G, H) 예측된 functional (G) SNV, (H) INDEL 수입니다. y축에 Log2값을 취하여 variant count의 비율을 표현했습니다.
위: 코호트 단계 비교
중간: 샘플 단계 비교
아래: FDR 비교
빨간 별은 샘플 NA12878에 100개 미만의 샘플이 있음을 의미합니다. (이 경우, FDR estimation의 신뢰도가 낮아집니다.)
(C), (D), (G), (H)의 FDR은 GIAB truth set v3.3.2.에 따라 sample NA12878을 대상으로 진행했습니다.
(A), (B), (E-H)는 상염색체만을, (C), (D)는 성염색체도 포함하여 분석했습니다.

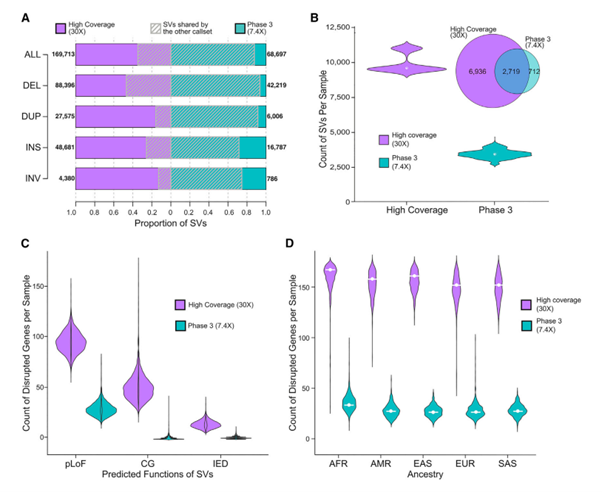
[Figure 4] ensemble SV calls와 phase 3 call set의 비교
(A) 서로의 structural variant call set가 얼마나 겹치는지 확인할 수 있는 그림입니다. SV에 관련된 각 데이터셋(ALL, DEL, DUP, INS, INV)의 SV count도 확인할 수 있습니다.
(B) 샘플 당 SV counts의 분포를 표현하며, 각 샘플 별 및 통합 call set에서의 겹치는 부분을 표현했습니다.
(C) 양쪽 데이터셋 모두에서 SV에 의해 변화한 gene의 수를 표현합니다.
pLOF(predicted loss of function); CG(complete copy gain); IED(intragenic exon duplication)
(D) Ancestral population 별로, SV에 의해 변화한 gene의 수를 표현합니다.

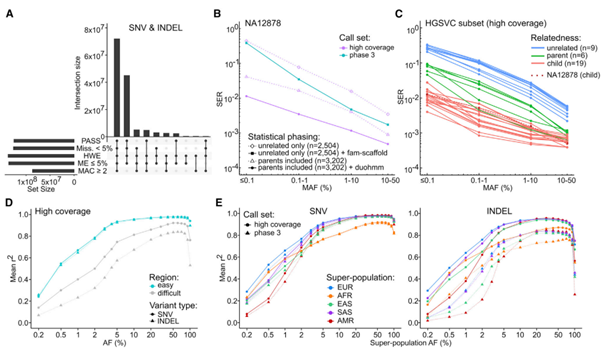
[Figure 5] Small variant 단계 생성 및 imputation 성능
(A) Small variants 중 조건(chr1-22, X; variant counts로 표현한 상위 10가지의
조건 조합을 나타냈습니다.)을 통과한 개수를 표현했습니다. (PASS:
VQSR을 통과한 sites; Miss.: genotype missingness;, HWE:
Hardy-Weinberg Equilibrium exact test, 1kGP super-population 중 최소 20%가 p-value > 1e-10인 small variants; MAC: Minor allele count)
위에서 ‘조건’이라 함은, haplotype phasing 과정에서 imputation에 사용할
수 있는 확실한 SNV/INDEL/SV call을 구성하는 데 reference
panel이 될 수 있는 데이터셋을 구성하기 위한 small variants set을 만드는
과정에 들어가는 일종의 quality control 입니다.
(B) High-coverage & phase 3 1kGP panel 간의
Haplotype phasing accuracy 비교값. (SER: switch
error rate – Platinum Genome truth set와 관련된 수치.) 두
가지의 추가적인 phasing condition(점선 표현 부분)은
평가 목적을 위한 high-coverage panel입니다.
(1) diamonds: NA12878 phasing 중 얻은 SER 수치. 코호트에 부모 정보가 없음.
(2) triangles: NA12878 phasing 중 얻은 SER 수치. 코호트에 부모님이 있지만, pedigree-based
correction(duohmm)이 적용되지 않은 데이터.
(C) High-coverage panel의 haplotype phasing accuracy. 샘플 간 연관성을 토대로 나누었습니다. HGSVC SNV call set와 연관지어 SER이 계산되었습니다.
(D) SNV, INDEL genotype의 imputation 정확도. High-coverage panel을 사용하여
imputation했으며, genomic region에 의해
나누었습니다. 110 SGDP 샘플에 비해 Mean r2,
squared Pearson correlation coefficient가 평균적으로 높았습니다.
(E) High-coverage 및 phase 3 panel 간의 imputation accuracy 비교입니다. Super-population ancestry에 의해 나누었습니다. 두
panel 간의 성능 비교는 공유되는 site에만 진행되었습니다.
(B-E)는 모두 상염색체 기준의 결과입니다.

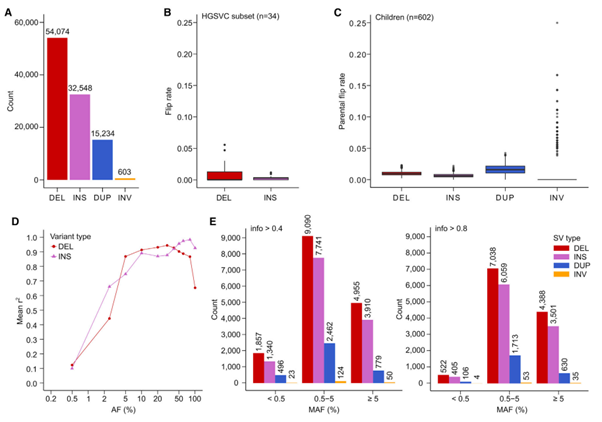
[Figure 6] SV phasing 및 imputation performance
(A) 통합 haplotype-resolved panel에 포함된 filtered SV의 코호트 단계 개수입니다. SV type에 의해 나뉘어 표현되었습니다.
(B) Sample-level phased HET DEL 및 INS에 대한 분포입니다. HGSVC truth set와 비교하여 phasing accuracy를 계산했습니다.
(C) Sample-level phased HET SV의 parental flip rate의 분포입니다.SV type으로 나누어 보았습니다.
(D) High-coverage panel의 SV imputation performance를 SGDP study dataset에 적용했으며, 분류는 SV type에 따라 나누었습니다. Imputed allele dosage 간 mean r2, squared Pearson correlation coefficient 값, 그리고 SV truth set로부터 온 dosage를 계산해 본 결과, 110 SGDP 샘플의 평균보다 높았습니다. (AF = 0.5% bin: 100 , 92개 샘플의 INS, DEL의 결과는 위 내용에서 제외입니다.)
(E) SGDP study dataset에서 imputation된 SV의 수를 표현합니다. 3개의 MAF bin에 대해 High-coverage reference panel은 다음과 같습니다. (왼: info > 0.4, 오: info > 0.8, 110개의 imputated SGDP sample 대상의 MAF)
(B-E): 상염색체 대상으로 진행했습니다.
Discussion
이 연구는 확장된 1kGP cohort의 high-coverage WGS의 결과를 제시하는데, 이는 2,504개의 원본 샘플뿐만 아니라 추가적인 698개의 관련 샘플로 구성되어 cohort에서 602개의 trio를 완성했습니다. 연구진들은 최첨단 방법을 사용하여 3,202개의 샘플에서 111,048,944개의 SNV, 14,435,076개의 INDEL 및 173,366개의 SV calling을 진행했습니다. 2015년의 low-coverage phase 3 1kGP dataset와 비교했을 때, high-coverage call set의 변형 수는 genome 당 추정 평균 190,885 SNV(1.05배), 268,182 INDEL(1.47배), 5,835(2.81배) SV의 증가, 그리고 cohort level에서 1,860만 SNV(1.24~9.05배), 기존 unrelated sample 2,504개에 걸쳐 최대 10만 SV(2.47배)의 증가를 보였습니다. 이들의 목표는 높은 coverage 및 phase 3 dataset 발견에 영향을 미칠 가능성이 있는 모든 요소를 해부하는 것보단, 기술 발전이 phase 3와 관련하여 high-coverage resource에 전체적으로 가져온 발전에 대한 광범위한 평가를 제공하는 것이었습니다.
예상대로, phase 3 dataset의 모집단의 거의 모든 공통 SNV(MAF > 1%)를 식별했다는 점을 감안할 때, high-coverage WGS에서 식별되었지만 phase 3에서는 식별되지 않은 SNV의 대부분은 희귀 MAF 스펙트럼(≤1%)에 속합니다. 또한, 여기서 발견된 대부분의 새로운 SNV는 low-coverage WGS 데이터 외에도 phase 3이 깊이 시퀀싱된 WES를 포함했다는 점에서 non-coding region입니다. High-coverage 시퀀싱과 현존하는 variant caller들이 SNV 호출에 비해 INDEL에 많은 개선을 가져온다는 사실과 맞게, 연구팀은 전체 MAF 스펙트럼에서 INDEL 카운트의 증가를 관찰했으며, rare end의 spectrum에서의 증가가 가장 두드러졌습니다.
여기에 제시된 SV는 phase 3 call set에 비해 discovery power이 크게 향상되었습니다. 이러한 데이터는 기술에 대한 직접적인 정밀 조사뿐만 아니라 inheritance의 성능 평가를 허용하는 family-based 설계를 통해 광범위한 algorithm 및 variant 평가의 이점을 갖습니다. 여기에 제공된 3.5%의 de novo SV 비율은 FDR을 위한 합리적, 혹은 불완전하다면 FDR을 위한 proxy입니다. 이 proxy에는 자식에게서 false positive SV, 정확히 예측된 true de novo variants SV(이전 단독 WGS 데이터 세트로부터 약 0.2%–0.5%로 추정[Collins et al., 2020; Turner et al., 2017; Werling et al., 2018]) 및 부모에서 false negative SV 또는 부모 또는 아이의 부정확한 breakpoint estimates가 포함됩니다. 또한 시간이 지남에 따라 셀 라인에서 발생하는 somatic variants를 포함하는데, 이는 위의 경우에 비해 낮을 것으로 예상되지만 그럼에도 불구하고 새로운 추정치에 변이의 일부에 기여할 것입니다. 특히 SV counts, size 및 frequency distributions, inheritance rates를 포함한 이 SV call set의 여러 특성은 혈액 유래 샘플에서 WGS에 이러한 방법을 활용하고, 새로운 SV 예측의 광범위한 분자 검증을 적용한 이전 연구와 비교할 수 있습니다(Werling et al., 2018). 우리는 모든 대형 CNV(>50kb, n = 4,180)에 대한 수동 검사를 수행하고 strand sequencing(Strand-seq)(>5kb, n = 250)에 대한 대규모 반전을 벤치마킹하여 직접적인 지원을 평가했습니다. 특히, 이 datatset에서 SV 발견에 제공한 중요한 발전은 각 human genome에서 SV의 기능적 변화에 대한 향상된 예측으로, 이는 phase 3 call set의 estimates(genome당 유전자 162개 : 32개)를 크게 능가하고 long-read WGS dataset(유전자 약 189개: Ebert et al., 2021)의 예측과도 유사합니다. 여기에 제시된 데이터는 HGSVC(Ebert et al., 2021)의 샘플 중 34개에 대한 독립적인 long-read WGS, Strand-seq 및 optical mapping datasets와 결합되어 방법 개발 및 게놈 연구를 위한 귀중한 open access SV 리소스를 제공합니다.
SNV/INDEL 및 SV 콜 세트 외에도, 향상된 SNV/INDEL 및 SV를 study datasets에 귀속하는 데 사용할 수 있는 개선된 haplotype-resolved reference imputation panel을 생성했습니다. 패널에 602개의 trio를 포함하면 관련 샘플 간의 long-range haplotype 공유의 증가와 phased haplotypes의 일관성을 보장하기 위해 통계적 단계 후 child sample에 적용되는 pedigree-based correction의 증가로 인해 panel 3에 비해 SNV/INDEL 단계의 정확도가 최대 10배 더 높아졌습니다.
HRC(The Happlotype Reference Consortium, 2016) 및 TOPMed(Taliun et al., 2021)와 같은 대부분의 기존 reference imputation panel들은 SV calling 및 phasing 문제로 인해 아직 SV를 포함하지 않습니다. 또한, SNV/INDEL에 대해 존재하는 Platinum Genomes(Eberle et al., 2017) 또는 GIAB(Wagner et al., 2022; Zook et al., 2019)와 유사하게 잘 확립된 haplotype-resolved SV truth set의 가용성으로 인해 SV phasing 정확도에 대한 평가가 어려웠습니다. 최근 발표된 HGSVC(Ebert et al., 2021)의 SV 통화 세트를 통해 후자의 문제를 회피할 수 있었고 SV phasing 정확도 평가에 필요한 참조를 제공했습니다. 또한 확장된 코호트에 trio가 포함됨에 따라 위상 정확도를 검증하는 직접적인 방식으로 inheritance pattern을 사용할 수 있습니다. 이러한 발전 덕분에 SNV/INDEL haplotype scaffold 위에 4개의 SV 타입(DEL, INS, DUP, INV)을 높은 정확도로 단계화할 수 있었습니다.
높은 신뢰도의 genotyped SV call set가 부족했던 탓에 high-coverage 1kGP panel의 SV imputation 성능을 평가하려고 시도할 때 문제가 되었습니다. HGSVC SV call set(Ebert et al., 2021)가 haplotype과 시퀀스 해결(long-read technology 의해 촉진됨) 둘 다라는 사실은 독립적인 SGDP 연구 dataset에서 SV “truth set”를 구축하기 위한 모집단의 구조적 변동 카탈로그로 사용할 수 있게 했습니다. 이러한 truth set으로 SV imputation 성능을 평가한 결과, imputed GT의 높은 정확도가 나타났으며, 이는 특히 MAF > 5%에서 small variants과 비슷했습니다.
근 10년 이상의 기간 동안, 1kGP 수집은 유전체학 분야의 핵심 자원이었습니다. 이러한 dataset은 인구 유전학과 게놈 생물학에 대한 과학적 통찰력을 제공했을 뿐만 아니라, 방법 개발 및 테스트에 더불어 기술적 검증에 널리 사용되는 공개적으로 공유 가능한 리소스를 제공했습니다. 완전한 phase 3 set에 대한 high-coverage sequencing data를 생성하고 추가 샘플로 602개의 trio를 완료함으로써, 연구진들은 차세대 대규모 국제 WGS initiative를 위한 벤치마크와 표준으로 해당 리소스를 업데이트했습니다. 이를 통해 더 높은 적용 범위와 시퀀싱 및 분석 방법의 발전은 frequency spectrum 전반에 걸쳐 모든 rare variants와 INDEL 및 SV의 발견을 크게 확대시켰습니다. Phase 3 set에 없는 많은 rare non-coding variants를 추가하면 코호트에 대한 다양한 유형의 모집단 유전자 연구가 가능할 것입니다. Pedigree correction을 활용하는 phased panel은 전반적으로 power를 향상시킬 것입니다. 특히 더 많은 일반적인 INDEL 및 SV의 imputation에 대한 개선을 제공하며, 이는 연관성 연구를 위한 imputation에 접근이 가능할 것입니다. 이 panel은 완전히 공개되어 있으며 다른 panel과 함께 자유롭게 다운로드하여 사용할 수 있고 모든 대상 dataset와 함께 사용할 수 있다는 점입니다. 현재 많은 대규모 시퀀싱 프로젝트가 수행되었지만, 1kGP 샘플의 개방성에 힘입어 향후 몇 년 동안 계속해서 커뮤니티의 표준 자원으로 남을 것입니다.