
11가지 tumour 유형에서 암으로 변화하는 과정의 epigenetic 조절

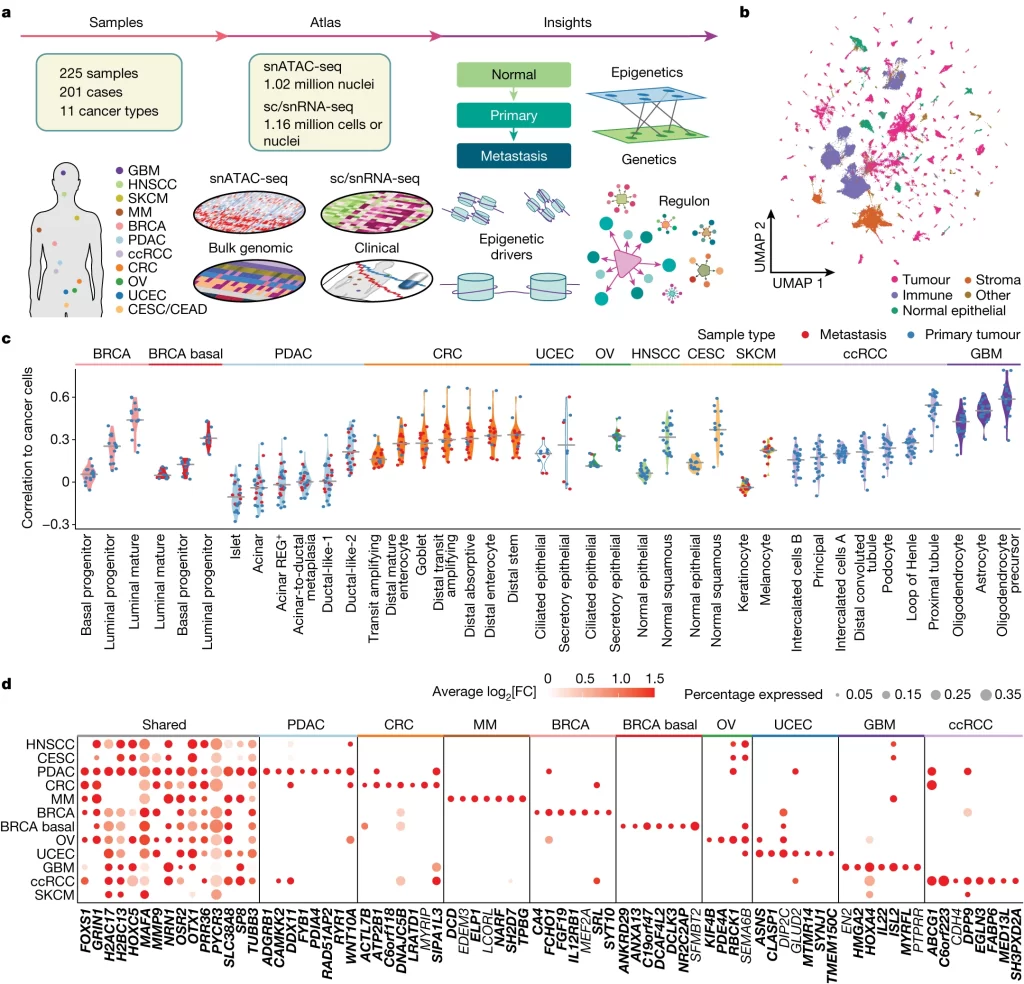
Abstract
Chromatin accessibility는 유전자 발현 조절 및 세포 정체성(Cellular identity)에 필수적이며, accessibility의 변화는 암의 시작, 진행 및 metastasis를 촉진하는 것과 관련이 있다. 발암성 전환(oncogenic transition)에 대한 genetic contribution이 연구돼왔지만, epigenetic driver에 대한 이해는 아직 부족하다. 여기서 우리는 225개 샘플의 *single-nucleus chromatin accessibility data (transposase-accessible chromatin에 대한 single-nucleus assay 사용)와 206개 샘플의 일치하는 single cell 또는 single-nucleus RNA sequencing expression data를 사용하여 pan-cancer의 epigenetics 및 transcriptomics 아틀라스를 구성했다. 각 플랫폼의 1억 개 이상의 세포를 1) 접근 가능한 chromatin 영역, 2) transcription factor motif 및 3) **regulons의 농축을 통해 분석한 결과, 우리는 암으로 전환되는 과정과 관련된 epigenetic drivers를 식별했다. 일부 epigenetic drivers는 여러 암에서 나타났고(예: ABCC1 및 VEGFA의 regulatory region; GATA6 및 FOX-family motifs), 나머지는 각 암에 특이적으로 나타났다(예: FGF19, ASAP2 및 EN1의 regulatory regions, PBX3 motif). epigenetic으로 변경된 경로 중 TP53, 저산소증(hypoxia) 및 TNF 신호 전달은 암 발생과 관련이 있는 반면, 에스트로겐 반응, 상피-중간엽 metastasis(Endothelial-Mesenchymal Transition, EMT) 및 apical junction은 암 Metastasis 과정으로의 변화와 관련이 있다. 또한, 우리는 enhancer accessibility과 유전자 발현 사이의 뚜렷한 상관관계를 밝혀냈고, epigenetic drivers와 genetic drivers 사이의 협력을 밝혀냈다. 이 아틀라스는 암으로 전환되는 과정의 epigenetic 역학에 대한 추가 조사를 위한 기반을 제공한다.
*single-nucleus chromatin accessibility data: 세포핵 내에서 chromatin에 접근이 가능한지 단일세포 레벨에서 분석한 데이터. DNA가 응축되지 않은 상태일 때는 Transposase가 접근해 분해가 가능하지만, Histone 단백질 등으로 인해 응축된 상태에서는 접근이 불가능한 차이를 이용해 chromatin accessibility를 확인.
**regulon: 같은 조절 단백질에 의해 조절되는 유전자의 집합
Figures
암 전반에 걸친 chromatin accessibility
– 11개 암 유형의 201명 환자로부터 225개 샘플 (158개의 primary tumour, 52개의 metastatic tumour, 15개의 *normal adjacent tissues (NATs))을 확보하고 멀티오믹스 데이터를 생성함.
*normal adjacent tissues: 암 조직과 인접한 정상 조직으로, 기본적으로는 정상 세포들로 구성됐지만 암 세포와의 직간접적인 상호작용으로 인해 다른 특징들을 가지고 있음.

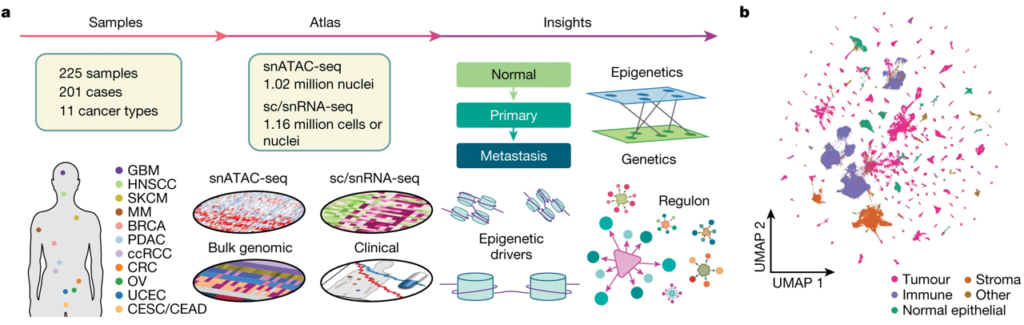
Fig. 1(A-B). 연구 설계 및 환자 코호트
(A) 수집된 암 및 샘플의 유형과 아틀라스의 구축·주석·통합, 그리고 생물학적 개체를 보여주는 데이터 생성 및 연구 설계 도식.
(B) 면역 세포 250,222개, 간질(stromal) 세포 69,684개, 정상 상피(normal epithelial) 세포 69,506개, 암 세포 588,895개의 분포를 보여주는 통합 pan-cancer snATAC-seq 결과의 UMAP(Uniform Manifold Approximation and Projection) 플롯.
tumour에서의 chromatin region 변화
– 정상세포에서 암세포로의 전환을 뒷받침하는 genetic/epigenetic 변화를 정의하고자 함
– chromatin accessibility 및 gene expression에 기반해 암세포와 가장 가까운 CNCs를 정의했고, CNC와 암세포를 비교해 조직 특이적 signal을 제거하고 조직 및 암세포 특이적인 *DACR (differentially accessible chromatin regions)을 식별
*DACR: 두 그룹에서 chromatin의 accessibility가 차이나는 region. RNA-seq의 DEG (Differentially Expressed Gene)와 비슷한 개념.

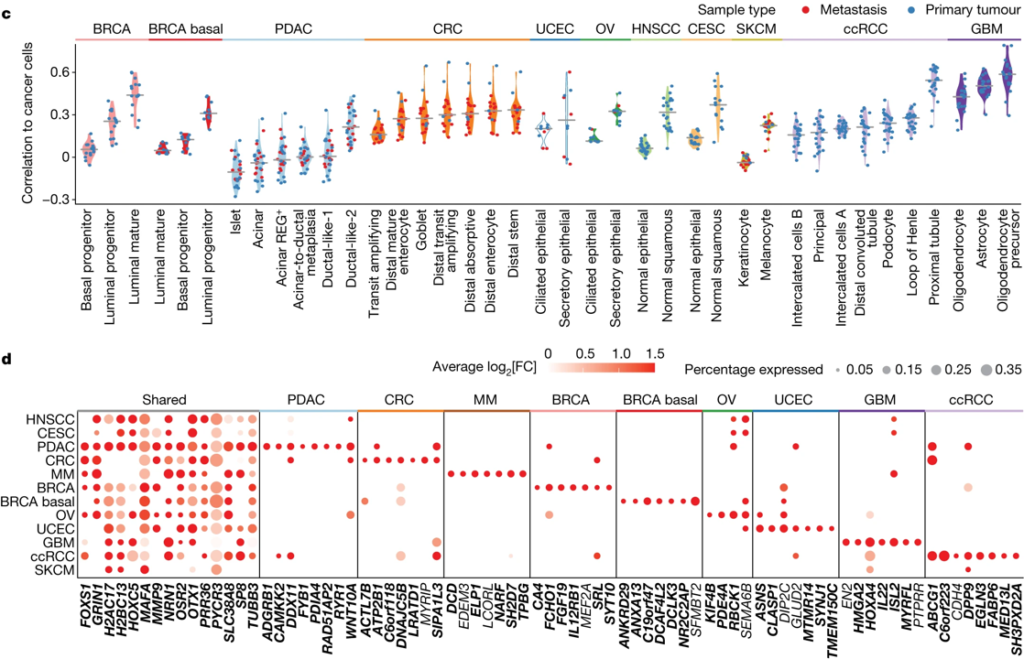
Fig. 1(C-D). 연구 설계 및 환자 코호트
(C) 각 1) tumour의 암세포와 2) tumour origin 조직의 정상 세포 유형 간의 chromatin accessibility로 Pearson 상관 계수를 계산. 세포 유형은 코호트당 중간 상관 계수가 큰 순서대로 정렬됨. 가장 오른쪽에 있는 세포 유형은 *CNCs (closest normal cells)로 간주되어 이후 암 관련 epigenetic driver를 식별하기 위한 reference로 사용.
(D) 암세포와 CNC를 비교하여 확인된 암세포 관련 top DACRs. 원의 크기는 접근 가능한 DACR이 있는 암세포의 비율을, 색상은 log2 Fold Change를 나타냄. x 축은 각 DACR에서 가장 가까운 유전자를 표시함.
*CNCs: 최근접 정상 세포. 본 연구에서 암 세포와 가장 correlation이 높은 정상 세포들로 정의됨.
[Fig 1C] chromatin accessibility가 암 세포와 가장 비슷한 정상 세포의 유형을 CNCs로 정의함. CNCs는 일종의 reference cell group으로, 암 세포가 가진 조직 특이성을 제외하고 암으로서 특이적으로 가지는 DACR을 분석하기 위해 사용됨.
[Fig 1D] 다양한 암종에 걸쳐 공유되는 DACR과, 각 암에 특이적으로 존재하는 DACR을 각각 보여줌.
tumour 진행 단계에서의 *ACR-gene link
– 전체적으로 enhancer 및 promoter region의 chromatin accessibility와 gene expression 사이의 연관 패턴을 분석
– Enhancer의 accessibility가 promoter에 비해 암의 유형 및 origin 조직에 더 특이적이었으며 transcripts expression도 더 잘 반영함.
– ACR-gene link를 갖는 chromatin region은 대부분 Enhancer에 해당
*ACR-gene link: 특정 지역의 chromatin accessibility와 gene expression간의 유의한 상관관계가 있는 경우 해당 관계를 ACR-gene link로 정의

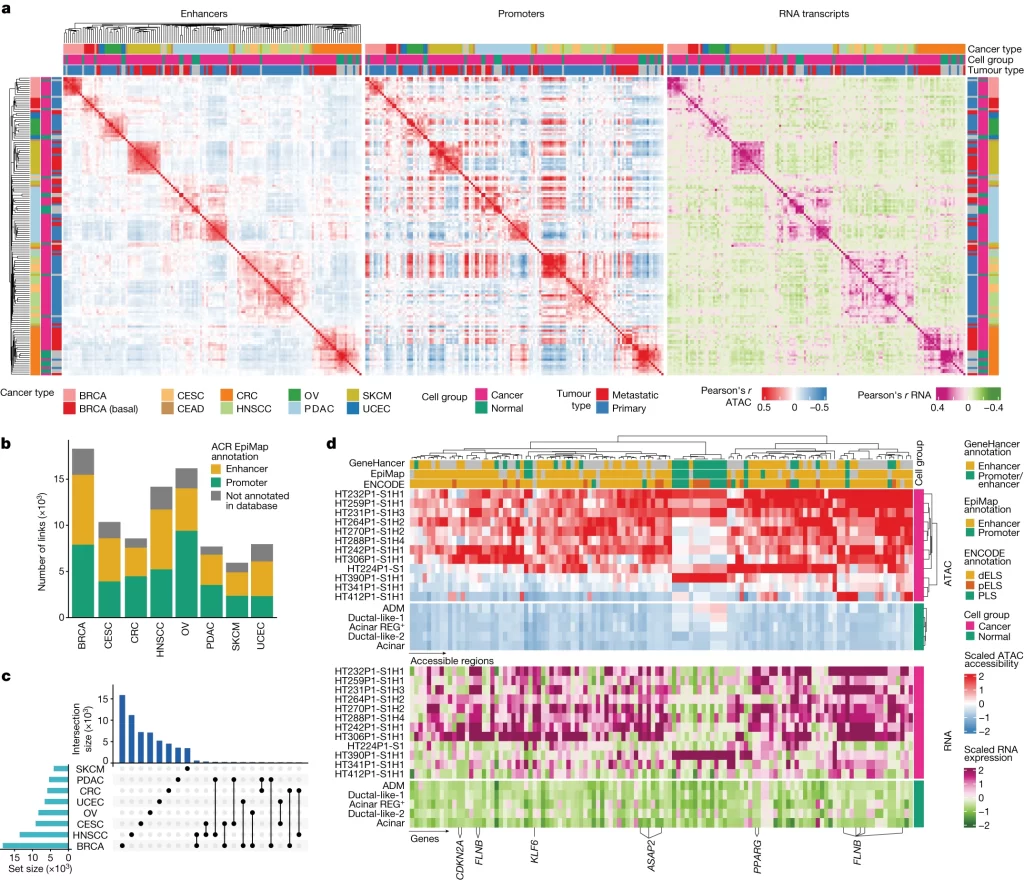
Fig. 2. 암의 전사 프로그램을 조절하는 CRE (Cis-Regulatory Element).
(A) 동일한 조직(*snMultiome-seq 샘플)의 암세포와 정상 세포의 샘플별 Pearson 상관 관계). 순서대로 (왼쪽) EpiMap enhancer 영역, (중앙) EpiMap promoter 영역, (오른쪽) RNA transcripts와 겹치는 ACR(accessible chromatin regions) 기반으로 계산됨.
(B) 암 유형별 ACR-gene link 수. 색깔은 ACR의 EpiMap annotation 기반 (Enhancer, Promoter, Not annotated).
(C) 대부분의 enhancer-gene link가 암 유형에 따라 다름을 보여주는 UpSet 플롯. 오른쪽 하단의 연결된 점은 표시된 암 유형 간에 공유되는 ACR-gene link을 나타냄.
(D) PDAC 암세포의 유전자 발현(아래)과 연관된 ACR(위)의 accessibility. Heatmap은 암세포의 경우 샘플별로, 정상 췌장 세포의 경우 세포 유형별로 집계된 average normalized & scaled된 snATAC-seq 및 snRNA-seq 값을 보여줌.
*snMultiome-seq: 하나의 nucleus에서 두 가지 이상의 paired omics data를 생산하는 sequencing 기법. 여기서는 chromatin accessibility와 gene expression을 paired data로 분석함.
[Fig 2A] promoter
region보다
enhancer에 해당하는
region에서 암
유형
및
origin 조직과 관련된 correlation 패턴이 잘
드러나고
있음.
이
패턴은
RNA transcripts에 해당하는
region으로 분석한 패턴과도 잘
일치함.
[Fig 2B-C] 유의한 ACR-gene link의
절반
가량이
유전자와
EpiMap enhancer 영역에 위치했으며, 대부분 각
암
유형에
특이적이었음.
[Fig 2D] ACR-gene link 중
primary tumour로의 전환
과정에
영향을
미치는
요소를
탐색.
대표적으로
PDAC에서 ASAP2의
encoding에 영향을
주는
3개의 enhancer가
유의한
link로
판별(이들의
promoter는 유의한
link가
없었음)됐는데,
이들은
기존
연구에서
focal adhesion을
활성화해
암세포의
proliferation을 돕는
기전이 알려짐.
Primary tumour의 regulon
– Metastasis가 이뤄지지 않은 primary tumor에서 조직 및 암세포 특이적 regulon을 조사함.
– 특정 TF의 motif accessibility 증가 및 그 타겟 유전자 그룹의 활성이 일치하는 주요 regulon들을 식별함.

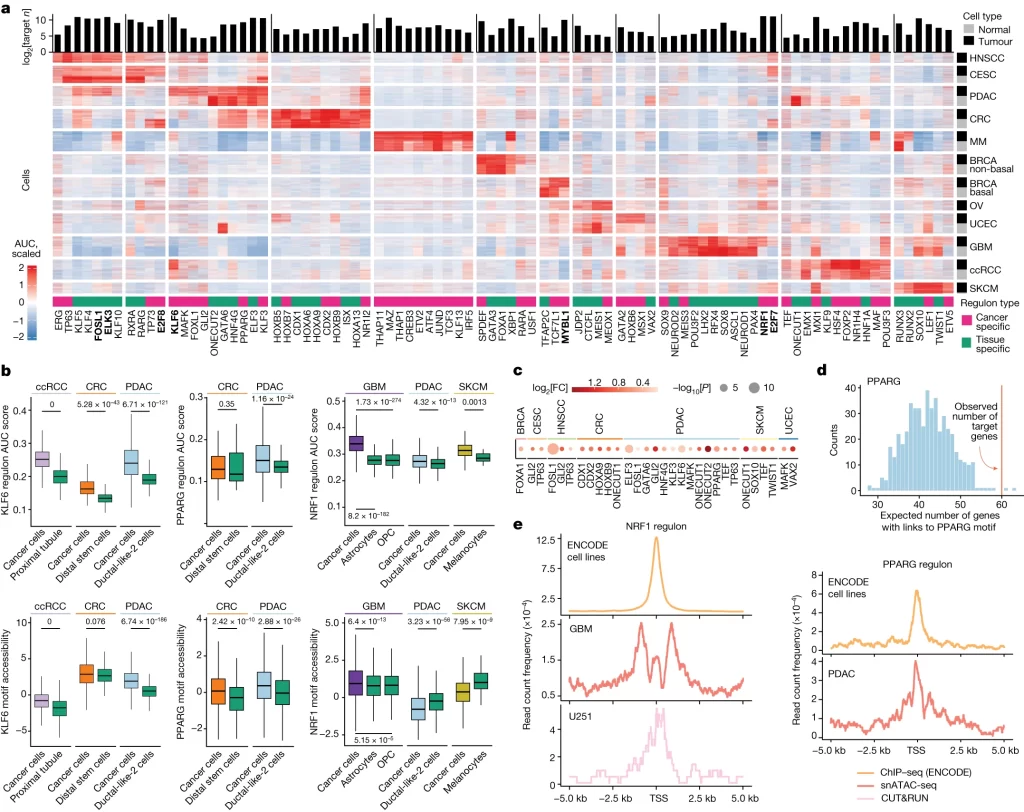
Fig. 3. pan-cancer 및 암 특이적 regulon.
(A) sc/snRNA-seq 데이터에서 *SCENIC을 사용하여 식별된 조직 및 암세포 특이적 regulon(열). 여기서 regulon은 TF와 이에 의해 조절되는 n개의 표적 유전자이며, 유전자 수가 맨 위에 표시됨. Heatmap은 각 암에서 무작위로 선택된 200개의 tumour 및 200개의 정상 세포(행)에 걸쳐 AUC(Area Under Curve) 점수를 표시. 암 특이적 regulon은 CNC에 비해 암세포에서 더 높은 활성을 나타내며, 여러 암에 걸쳐 공유되는 상위 암세포 특이적 regulon은 굵게 강조 표시.
(B) Primary 암 세포 및 해당 CNC의 regulon activity score (상단) 및 TF motif accessibility score (하단). 색상은 각 암 유형을 나타내며, 녹색 상자는 정상 세포를 나타냄.
(C) TF 특이적 ACR-gene link(ACR이 해당 TF의 binding site를 포함)에 대해 표적 유전자가 enriched된 TF. 색상은 이러한 링크가 있는 무작위 유전자의 예상 수(K1, …, n)에 대한 이러한 링크가 있는 관찰된 표적 유전자 수 사이의 log2 Fold Change를 나타냄. 단측 P-value는 Gaussian Z score로 각 regulon에 대해 계산.
(D) 무작위로 샘플링된 유전자에서 발견된 PPARG 특이적 PDAC ACR-gene link을 갖는 유전자 수의 정규 분포의 예. PPARG 특이적 ACR-gene link이 있는 관찰된 PPARG 표적 유전자의 수는 빨간색 선으로 표시.
(E) 표적 유전자의 TSS (transcription start site) 주변에 ChIP-seq 피크(ENCODE), snATAC-seq 피크 또는 CUT&RUN 피크의 존재.
* SCENIC (Single-Cell rEgulatory Network Inference and Clustering): 유전자간의 상호작용을 네트워크로 분석하는 방법으로, 각 TF에 의해 조절되는 유전자군을 식별하는 데 사용됨
[Fig 3A] CNC 대비 여러 regulon이 각 암종 특이적이거나 공통적으로 tumour cell에서 활성이 증가함.
[Fig 3B] 암종 하나에 대해 활성의 차이를 보이는 regulon들을 나타냄. 예를 들어 KLF6의 regulon은 ccRCC, CRC, PDAC에서 활성이 증가하고, motif accessibility도 증가했음. 이는 KLF6의 작용이 강화되고 그 하위 유전자 그룹의 활성이 증가하는 일관된 증거임.
[Fig 3C-D] 21개의 TF가 random gene에 비해 각 TF의 binding site를 포함하는 ACR과 더 link되는 경향이 있음을 보여줌.
[Fig 3E] TF가 실제로 해당 regulon에 속한 하위 유전자들의 transcription start site에 결합하고 있음(TF의 표적 유전자가 맞음)을 보여줌.
암 metastasis의 epigenetic 프로그램
– Primary, metastasis sample이 paired된 case를 이용해 metastasis를 유발하는 epigenetic 변화를 확인함/
– TF motif accessibility scores를 계산해 EMT 등 중요한 형질 변화의 master regulator를 찾고 regulon의 변화로 검증함.
– Enriched pathway 및 trajectory 분석을 통해 metastasis 과정에서 암별로 활성화되는 pathway를 확인하고, normal cell부터 primary-metastasis 단계로 변화하는 과정을 분석함

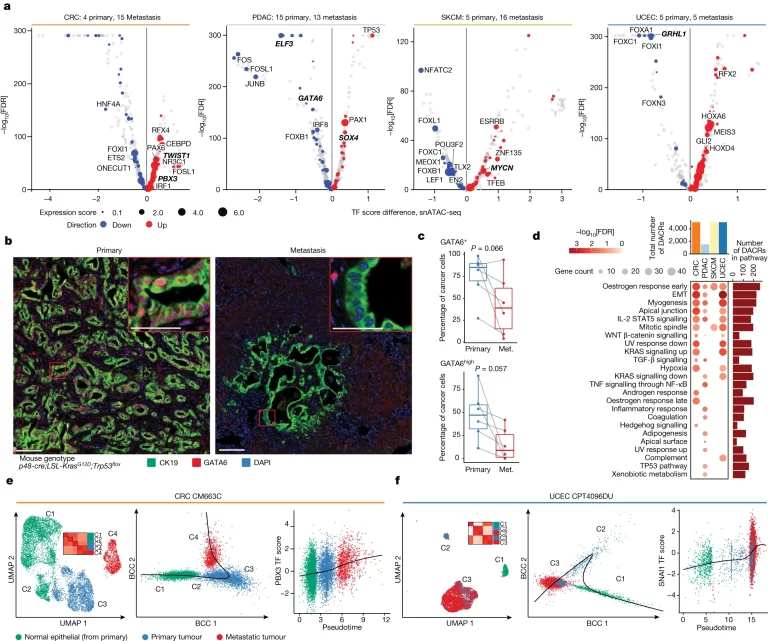
Fig. 4. 암 metastasis에서 활성화된 epigenetic 프로그램.
(A) 4가지 암 유형에서 metastasis와 primary sample 샘플 사이 motif accessibility이 차이나는 TF. y축은 motif accessibility의 유의한 차이 여부를, x축은 TF expression의 차이 정도를 보여줌 (log2 fold change)
(B) PDAC의 마우스 모델에서 GATA6(빨간색) 발현에 대한 *mpIHC 분석. CK19(녹색)는 암세포, DAPI(파란색)는 핵을 표시.
(C) GATA6+ 및 GATA6high 암세포 비율은 매칭된 metastatic(met.) PDAC에 비해 primary PDAC에서 더 높았음.
(D) metastatic tumour 및 primary tumour에서 상향 조절된 DACR의 유의하고 암시적인(FDR ≤ 20%) hallmark pathway enrichments. 원의 크기 및 색상은 각각 유전자 수와 log10(FDR) 값을 표시. 분석에 사용된 암 유형별 총 DACR 수는 log2 Fold Change 기준 상위 5,000개로 제한(상단). 각 pathway로 annotated된 총 DACR 수가 오른쪽에 표시.
(E,F) CRC 사례(E)와 UCEC 사례(F)의 primary 및 metastasis 샘플 쌍에 대한 UMAP 플롯(왼쪽). 작은 Heatmap은 각 샘플의 클러스터당 평균 TF-모티프 점수를 기반으로 하는 Pearson의 상관계수를 보여줌. (중앙) Slingshot으로 식별된 궤적을 따라 정렬된 셀을 보여주는 산점도와 (오른쪽) PBX3 (E) 또는 SNAI1(F) motif accessibility와 pseudotime의 진행사이의 연관성을 보여주는 산점도.
*mpIHC 분석: Multiplex Immunohistochemistry, 생체 조직 샘플에서 여러 종류의 단백질을 동시에 검출하고 시각화하는 방법
[Fig 4A] x축에서
y축은 two-sided Wilcoxon rank sum test를
사용하여
계산된
FDR corrected P value를
보여줌.
Expression score는
metastasis와 primary cancer cell TF 발현량
사이의
log2 fold change의
절대값을
계산한
후,
샘플별로
평균값을
취함.
동일한
TF-cancer pair에
대한
motif score 차이와 동일한
Fold change 방향을 계산함.
[Fig 4B-C] GATA6는 primary PDAC에서
풍부했지만
metastasis 과정으로
변화하며
감소하는
주요
regulator임.
[Fig 4D] EMT,
myogenesis and apical junction 등이
3개
cohort에서 metastasis와 연관된
pathway였으며,
각
암종에서
특이적으로
강화되는
pathway들도 존재함.
[Fig 4E-F]
pseudotime에 따라 각 각 암종에서 normal-primary
tumour-metastatic tumour로 진화하는 과정을 모델링.
genetic & epigenetic 상호작용 및 임상적으로 연관된 epigenetic 프로그램
– Genetic driver (DNA mutation)과 epigenetic (chromatin accessibility) 사이의 상호작용을 조사
– Mutation이 자주 일어나는 TERTp region에서 mutant allele이 있는 경우 chromatin accessibility가 차등적으로 높았음
– oncogene으로 알려진 유전자들의 Enhancer에 해당하는 region에서 ACR-gene expression link를 조사
– regulon 활성화에 따른 예후 차이 분석 및 인간유두종바이러스(HPV)와 TF 활성화 관계 조사

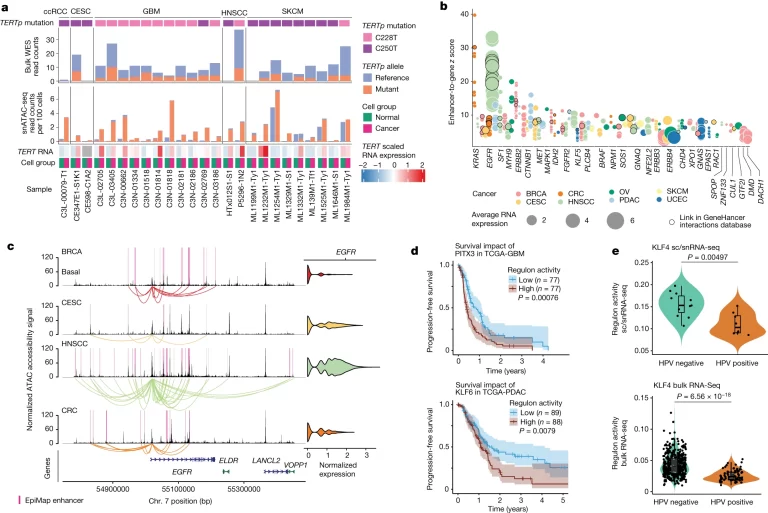
Fig. 5. genetic driver가 chromatin accessibility에 미치는 영향.
(A) snATAC-seq 데이터 및 WES 데이터로부터 5가지 암 유형에서 검출된 TERTp 돌연변이(C228T 및 C250T). 대량 WES 데이터(상단) 및 snATAC-seq 데이터(하단)에서 레퍼런스 또는 돌연변이 대립유전자에 해당하는 read counts가 표시됨. snATAC-seq 지원 읽기는 암세포와 정상 세포에 대해 별도로 계산된 다음 각 그룹의 총 세포 수로 정규화. 하단의 Heatmap은 샘플당 암 및 정상 세포의 TERT 발현을 보여줌.
(B) snMultiome-seq 데이터를 사용하여 확인된 알려진 tumour유전자의 epigenetic적 조절. 각 점은 하나의 enhancer-gene link의 z-score를 나타냄. enhancer-gene z-score는 EpiMap 또는 GeneHancer 데이터베이스에 annotation된대로 하나의 enhancer에 속하는 모든 ACR에 대한 ACR-gene link z-score의 평균으로 계산. 점 색상은 ACR-gene link이 확인된 암 유형을, 점 크기는 x 축에 표시된 유전자의 normalized RNA expression을 나타냄.
(C) BRCA basal, CESC, HNSCC 및 CRC 암세포에서 EGFR 영역의 적용 범위 플롯. neutral EGFR CNV가 있는 샘플만 포함. EGFR RNA expression은 오른쪽에 표시.
(D) PITX3 regulon 활성에 의해 계층화된 TCGA-GBM 코호트(상단) 및 KLF6 regulon 활성에 의해 계층화된 TCGA-PDAC 코호트(하단)에서의 Kaplan-Meier 플롯 및 무진행 생존 분석.
(E) 본 연구의 HPV 양성 및 HPV 음성 HNSCC 샘플(상단) 및 TCGA-HNSCC(하단)에서 KLF4의 Regulon 활성.
[Fig 5A] TERTp mutant allele에 대한 chromatin accessibility가 cancer cell에서 유의하게 높았음. 이는 TERT의 expression이 cancer cell에서 더 많아지는 원인임.
[Fig 5C] 30 oncogenes of which the expression was linked to enhancer accessibility, with the strongest links in EGFR, KRAS, ERBB2, CTNNB1 and MET 30개의 oncogene expression이 enhancer 부위에 대한 accessibility와 linked됨. 가장 강한 link로는 EGFR, KRAS, ERBB2, CTNNB1 등이 있음.
[Fig 5D] 각 regulon의 활성에 따라 두 환자그룹의 예후에 유의한 차이가 있음을 보여주는 그림.
[Fig 5E] 인간 유두종 바이러스(HPV) 상태가 활성 TF의 환경에 미치는 영향을 추가로 조사한 결과, HPV- tumour과 비교하여 HPV+ tumour에서 KLF4 레굴론 활성의 현저한 감소를 관찰.
Disscussion
우리는 11개 암 유형에 걸쳐 225개 샘플에서 tumour 및 NAT의 대규모 단일 세포 multi-omics 아틀라스를 만들고 조사하여 다양한 암 및 정상 조직 세포 유형을 공개했다. 이전의 대량 ATAC/RNA-seq 연구를 뛰어넘어 우리의 분석은 암 특이적 epigenetic 구조, 정상 세포와 악성 세포 사이의 관계, 동일한 계통의 primary-to-metastasis로의 전환을 포함하여 암 생물학에 대한 섬세한 통찰력을 제공한다. 우리는 암세포와 공유되는 chromatin accessibility 패턴을 기반으로 CNC 유형을 식별했다. 이는 세포 계통을 암시할 수 있으며, 원발 세포를 결정하는 데 있어서의 epigenetic 구조의 중요성을 강조한다. 뿐만 아니라 정상 세포가 암으로 전환되는 과정에 중요한 통찰력을 제공한다. 동일한 유형의 primary 암과 metastatic 암 사이의 chromatin accessibility 변화를 확인하고 tumour 유형을 비교함으로써 암 유형 전반에 걸쳐 암 진행을 관리하는 chromatin 환경 및 epigenetic 프로그램의 공통점 및 차이점이 모두 강조되었다.
primary tumour와 metastatic tumour 사이의 chromatin accessibility 차이는 metastasis를 방해하는 방법을 암시할 수 있다. GATA6 TF 모티프는 개방된 chromatin에서 고갈되고, GATA6 발현은 primary PDAC에 비해 metastatic PDAC에서 감소한다. 또한, GATA6 regulon 활성은 각각의 CNC와 비교하여 PDAC, CRC, OV 및 UCEC primary 암 세포에서 감소한다. PDAC의 GATA6 손실은 기존 발견과 일치하게 EMT 표현형을 유도하고 basal subtype과 관련이 있으며, 전체 생존율을 감소시킨다. 우리는 이전에 보고되지 않은 많은 추정 epigenetic driver와 중요한 TF와 상관관계가 있는 cis regulatory element를 확인했다. 우리는 또한 in vitro 및 PDAC xenograft 모델에서 세포 이동과 tumour 성장을 촉진하는 primary PDAC 세포의 ASAP2 상향 조절과 연결된 proximal 및 distal enhancer를 식별했을 뿐만 아니라 세포 운동성, 침입 및 증식에 관여하는 유전자(GDF15 및 FGD3)들의 chromatin accessibility에 영향을 미치는 genetic driver (BRCA의 TP53 돌연변이)를 식별했다. 이 연구에서 bulk WES, snRNA-seq 및 snATAC-seq의 통합 분석은 암 전반에 걸쳐 TERTp 돌연변이의 대립유전자 특이적 chromatin accessibility 효과를 더욱 강조한다. EGFR, KRAS 및 MET와 같은 cancer drivers와 관련 enhancer 사이의 강력한 연관성 발견은 tumour 형성 중 epigenetic 및 genetic 상호 작용의 중요성을 더욱 강조한다. 추가 기능 검증을 통해 암의 주요 regulatory pathway가 밝혀질 것이다.
tumour 전체의 chromatin 구조, 암으로의 핵심적인 변화 시 chromatin accessibility 변화, chromatin accessibility와 genetic alteration 및 transcription 패턴 간의 상호 작용을 이해하는 것은 암 생물학 및 임상 실습을 발전시키는 데 중요하다. 암 발생 및 metastatic 확산의 중요한 사건/driver을 나타내는 chromatin accessibility의 특정 변화는 잠재적인 치료 표적이 될 수 있다. TF 자체는 전통적인 치료법으로 표적화하기가 매우 어렵고 정상 조직에서의 다양한 역할로 인해 표적 외 효과에 대한 우려가 제기되지만, 우리는 광범위한 전사 프로그램에 초점을 맞춤으로써 잠재적으로 표적화 가능한 요소를 강조했다. 마지막으로, 우리는 이 아틀라스가 미래의 암 연구를 위한 귀중한 자원이 될 것으로 기대한다.