
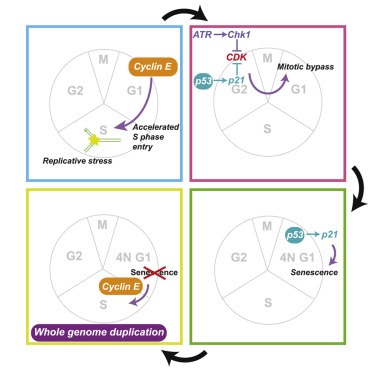
Cyclin E-induced replicative stress는 p53-dependent whole-genome duplication을 유도한다.

Abstract
Whole-genome duplication (WGD)는 암 진화에서 자주 발생하는 사건이며 신경계의 중요한 driver이다. WGD에서의 p53 tumor suppressor의 역할은 수수께끼 같았다. p53은 tetraploid cell의 증식을 차단하여 WGD의 장벽으로 작용할 수 있지만, endoreduplication를 통해 WGD의 핵심 단계인 mitotic bypass를 촉진할 수도 있다. wild-type p53 tumors에서 WGD는 종종 E2F 경로의 활성화, 특히 cyclin E1을 암호화하는 CCNE1의 amplification과 관련이 있다.
여기서 우리는 상승된 cyclin E1 발현이 ATR 및 Chk1-dependent G2 phase arrest를 활성화하는 replicative stress를 유발한다는 것을 보여준다. p53은 Wee1과 함께 downstream 표적 p21을 통해 mitotic cyclin-dependent kinase activity를 충분히 억제하여 APC/CCdh1을 활성화하고 mitotic bypass를 촉진한다. Cyclin E 발현은 mitotic bypass 후 p53-dependent senescence를 억제하여 세포가 endoreduplication를 완료할 수 있게 한다. 우리의 결과는 p53이 WGD의 촉진을 통해 암 진화에 기여할 수 있음을 나타낸다.
Figure

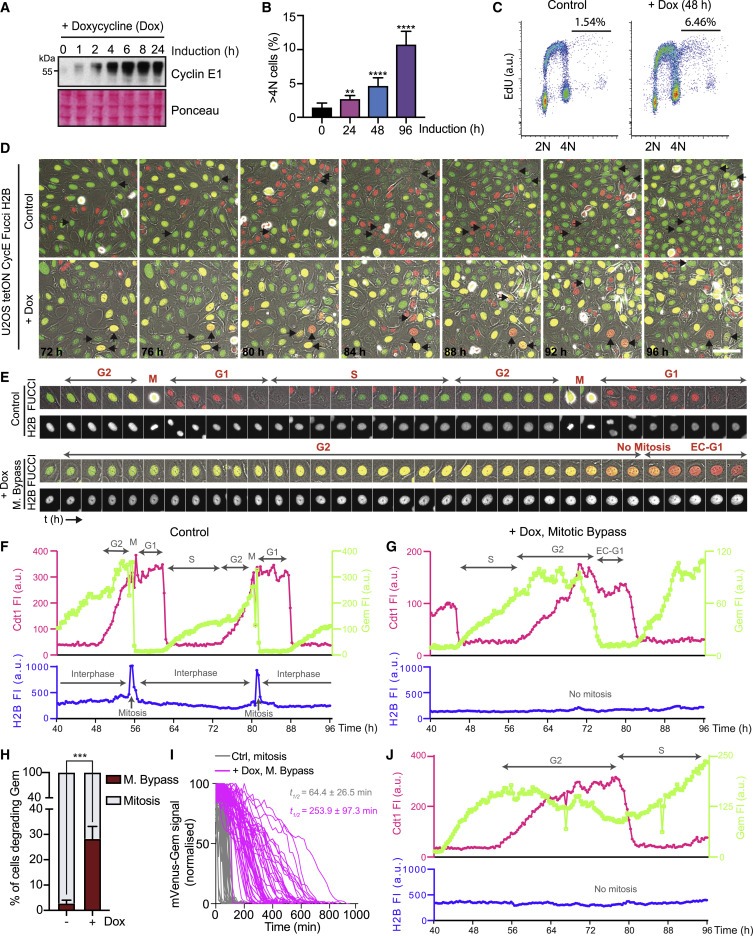
Figure 1. cyclin E 발현은 U2OS 세포에서 endoreduplication를 유도한다.
(A) U2OS tetON CycE cell line에서 doxycycline (Dox) 처리에 의한 cyclin E1(CycE) 발현을 나타내는 Immunoblots.
(B) total cell의 %로 측정된 U2OS에서 96시간 동안 cyclin E 유도 후 >4N DNA 함량을 갖는 세포의 Quantification.
(C) cyclin E 유도 후 EdU를 처리한 U2OS 셀의 FACS 분석.
(D 및 E) cyclin E. FUCCI를 발현하는 U2OS 세포의 Time-lapse imaging와 phase contrast image 병합.
(F 및 G) cells. a.u., arbitrary unit의 mCherry-cdt1 및 mVenus-Gem의 fluorescence intensities (FI)에 대한 Temporal profile.
(H) geminin을 분해한 세포의 %(S/G2를 완료하는 세포의 %)에서 측정된 mitotic bypass의 Quantification.
(I) mVenus-Gem의 Normalized time-course degradation.
(J) 높은 수준의 mVenus-Gem을 유지하면서 mCherry-Cdt1을 저하시킨 cyclin E-발현 U2OS cell의 Temporal profile.

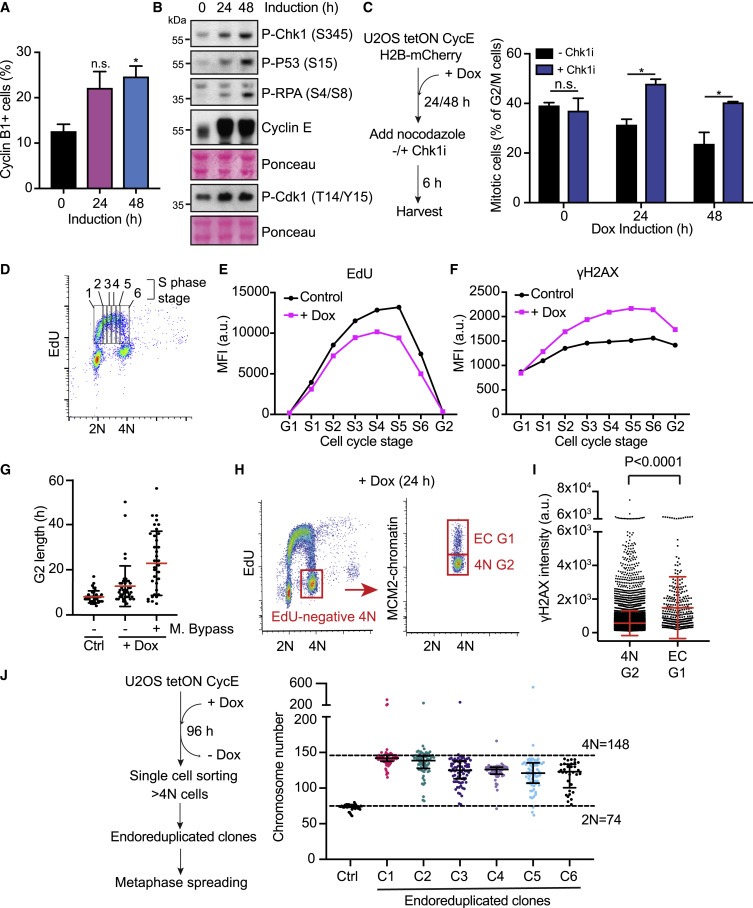
Figure 2. cyclin E-expressing cells의 Replicative stress.
(A) FACS 분석에 의한 cyclin 유도 후 cyclin B1에 양성인 U2OS 셀의 Quantification.
(B) U2OS 세포에서 cyclin E 유도 후 DNA damage marker를 보여주는 Immunoblots.
(C) 왼쪽: 실험 접근 방식의 도식
오른쪽: mitotic cells의 평균 percentages.
(D-F) cyclin E-발현 세포의 Replicative stress.
(G) 그림 1D의 대표적인 FUCCI 실험의 개별 세포의 G2 길이.
(H 및 I) 24-h cyclin E 유도 후, EC-G1 및 4N G2 모집단에 대한 FACS 분석(H). 대표적인 실험에서 개별 세포에 대한 γH2AX 수준 (I).
(J) endoreduplicated 클론의 Karyotypes.

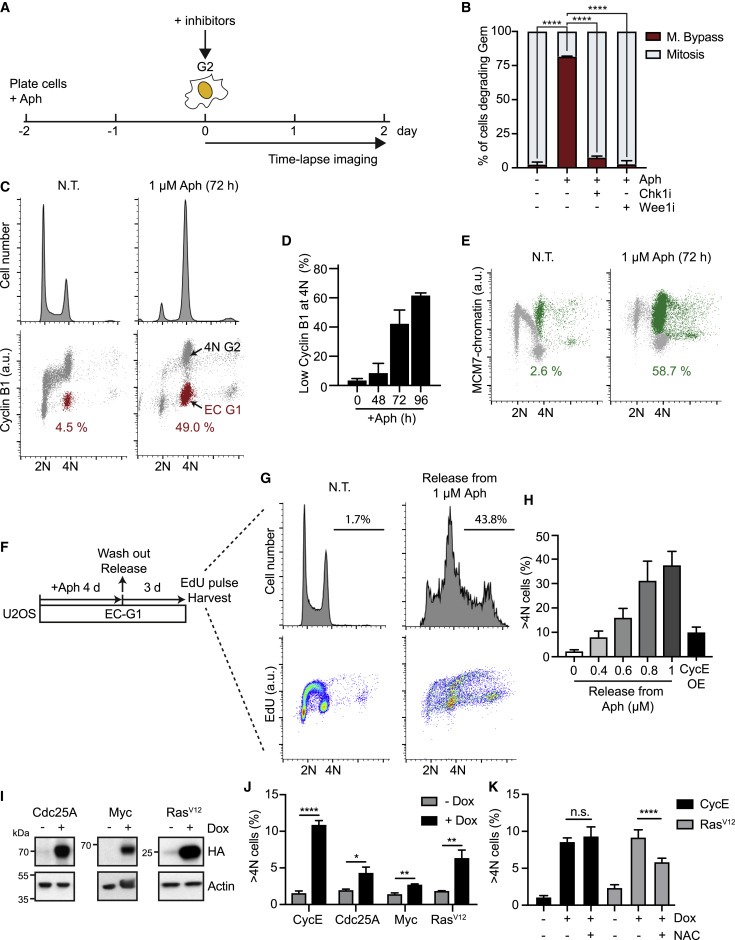
Figure 3. aphidicolin 처리 및 oncogene-expressing cell의 mitotic bypass.
(A 및 B) 실험 방법의 개략도(A) geminin을 분해한 세포의 %(S/G2를 완료하는 세포의 %)에서 측정된 mitotic bypass의 Quantification(B).
(C-E) 1 μM aphidicolin (Aph)으로 72시간 동안 처리된 U2OS 세포는 DNA 함량, cyclin B1 수준, chromatin-bound MCM7 수준 분석(C 및 E), EC-G1 세포의 평균 percentage (D).
(F-H) 실험 방법의 개략도 (F), 방출된 세포를 DNA 함량 및 DNA 합성(EdU)(G)에 대해 분석 (G). EdU를 포함하는 >4N 세포의 정량화 (H).
(I) U2OS tetON cell lines에서 Dox 처리에 의한 Cdc25A, Myc 또는 RasV12 유도를 보여주는 Immunoblots.
(J) cyclin E(CycE), Cdc25A, Myc 또는 RasV12의 96-h 유도(Dox) 후 FACS 분석에 의한 4N 이상의 DNA 함량을 가진 U2OS 세포의 Quantification.
(K) CycE 또는 RasV12(+Dox)를 발현하도록 유도된 U2OS 세포는 5mM N-acetyl cysteine (NAC)으로 배양되었다. FACS 분석에 의한 4N 이상의 DNA 함량을 가진 세포의 Quantification.

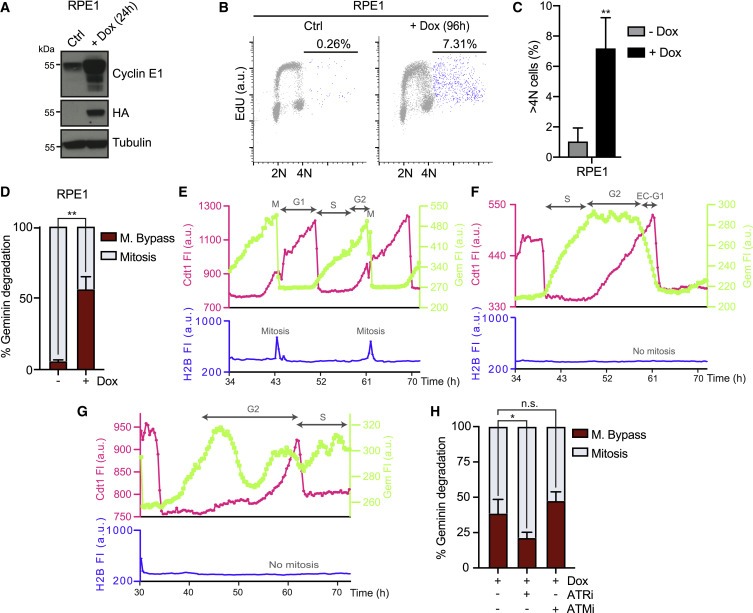
Figure 4. Cyclin E 발현은 RPE1 세포에서 endoreduplication를 유도한다.
(A) RPE1 tetON CycE cells에서 cyclin E 유도(+Dox)를 나타내는 Immunoblots.
(B) 96-h cyclin E 발현 후 EdU를 포함하는 RPE1 세포의 FACS 분석.
(C) (B) SD(n = 4)에서 전체 세포의 %로 측정된 4N 이상의 DNA 함량을 가진 세포의 Quantification.
(D) geminin을 분해한 세포의 %(S/G2를 완료하는 세포의 %)에서 측정된 mitotic bypass의 Quantification.
(E 및 F) mitosis을 bypassing하는 대조군 RPE1 세포와 Dox 처리된 RPE1 세포의 mCherry-cdt1 및 mVenus-Gem의 fluorescence intensities (FI)에 대한 Temporal profiles.
(G) 높은 수준의 mVenus-Gem으로 mCherry-cdt1을 분해하는 Dox 처리된 RPE1 세포의 Temporal profiles.
(H) SD로 mitosis를 bypassed한 RPE1 세포의 평균 percentages.

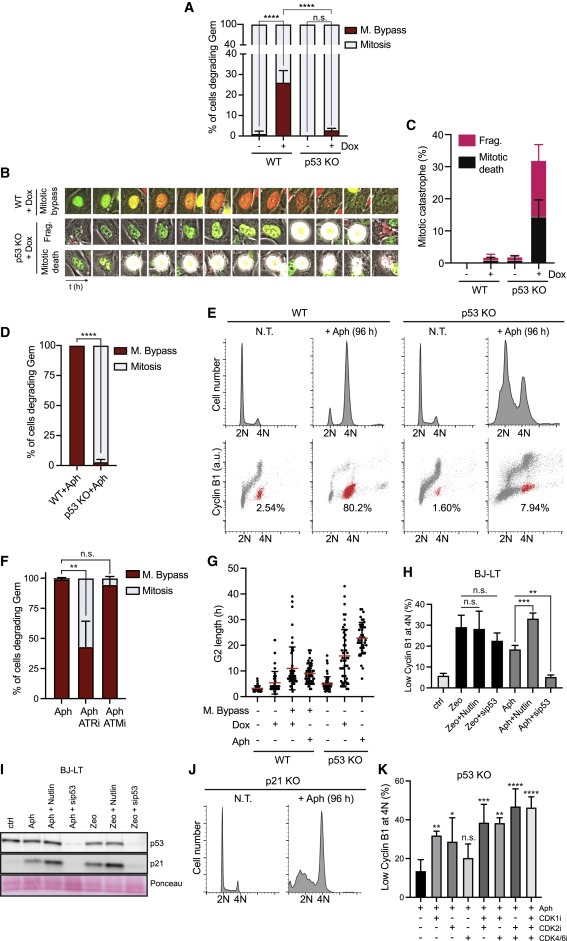
Figure 5. p53 녹아웃은 aphidicolin-treated 및 cyclin E-발현 RPE1 세포에서 mitotic bypass를 폐지한다.
(A-C) RPE1 WT 및 p53 KO 세포를 유도하여 cyclin E(+Dox)를 발현시키고 96시간 동안 촬영하였다. Mitosis를 SD로 bypassed한 세포의 Quantification (A), 예제 셀의 Selected images (B), nuclear fragmentation (Frag.)와 mitotic death의 Quantification (C).
(D) RPE1 WT 및 p53 KO 세포를 1 μM의 aphidicolin (Aph)으로 처리하고 72시간 동안 영상화하였다(Video S5 참조). SD를 사용한 mitotic bypass의 Quantification
(E) 1 μM Ph로 처리된 RPE1 WT와 p53 KO 세포를 FACS로 96시간에 분석하였으며, EC-G1 세포를 식별하여 빨간색으로 표시.
(F) SDs로 mitosis를 bypassed한 RPE1 세포의 Quantification
(G) single representative experiment를 통한 (A)와 (D)에서 세포의 G2 length 측정.
(H 및 I) 0.5 μM aphidicolin (Aph) 또는 50 μg/ml zeocin (Zeo)으로 72시간 동안 처리된 BJ-LT 세포를 2 μM Nutlin(24시간) 또는 p53 siRNA(0시간)로 보충한 후 immunoblots과 FACS 분석. p53 및 p21 발현을 나타내는 immunoblots (I), 4N DNA 함량에서 낮은 cyclin B1을 가진 세포의 평균 percentages (H).
(J) 1μMAh로 처리된 RPE1 p21 KO 세포의 FACS를 이용한 DNA content 분석.
(K) 48시간 동안 1μM Ph로 처리된 RPE1 p53KO 세포에 FACS 분석 전 24시간 동안 CDK1, CDK2 또는 CDK4/6의 inhibitors를 추가로 보충하였다. 4N DNA 함량에서 낮은 cyclin B1을 가진 세포의 평균 percentages.

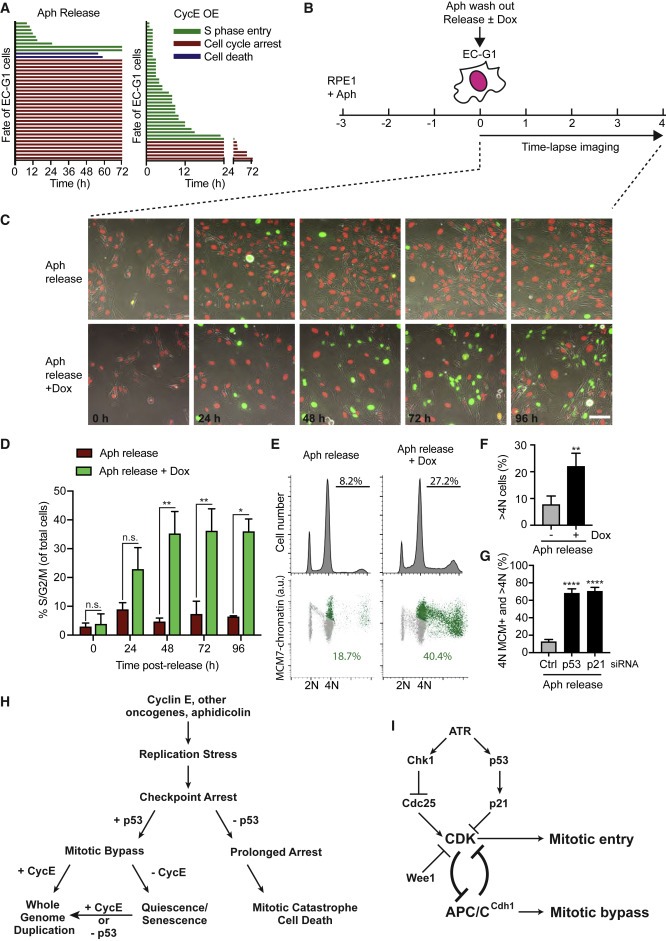
Figure 6. CycE 발현은 노화된 EC-G1 세포의 endoreduplication를 재정립한다.
(A) 0.5 μM의 aphidicolin (Aph) 처리로부터 방출되거나 CycE 발현으로 생성된 RPE1 EC-G1 Individual cell fate.
(B-D) 실험 방법의 개략도 (B), 0.5 μM Ph를 사용하였다. 지정된 시점의 Still image (C), 지정된 시점에서 S/G2/M phase에 있는 세포의 SEM을 사용한 평균 percentage (D)
(E 및 F) RPE1 세포는 (B)와 같이 처리되었고, DNA 함량 및 MCM 로딩을 위해 Ph 방출 후 96h에서 FACS에 의해 분석되었다. EdU를 포함하는 >4N 세포의 Quantification (F)
(G) 세포는 (B)와 같이 처리되고 96시간 동안 siRNA로 방출되었다. 4N DNA 함량 또는 >4N DNA 함량에서 높은 MCM 수준을 가진 세포의 SD를 사용한 평균 percentage.
(H 및 I) p53 -positive cell에서 oncogene-induced replicative stress에 의해 구동되는 whole-genome duplication 모델.
Disscussion
그림 6H와 6I에 요약된 우리의 결과는 p53이 필요한 endoreduplication를 통한 WGD의 경로를 설명한다. 이 경로는 oncogene 발현의 일반적인 결과인 replicative stress의 생성으로 시작된다. cyclin E 발현의 경우, replicative stress는 G1 단계 단축으로 인한 replication origin licensing system의 perturbation에서 발생한다(Macheret and Halazonetis, 2018; 17 Matson et al., 2017; 18 및 본 연구). 그러나 RasV12는 origin licensing을 감소시키지 않는다(그림 S3H). 대신 reactive oxygen species의 생성을 통해 replicative stress를 생성한다. 우리의 결과는 두 가지 유형의 스트레스가 mitotic bypass와 WGD를 유도할 수 있음을 보여준다. 게다가, aphidicolin 과 같은 replicative stress의 exogenous source는 mitotic bypass를 유도할 수 있다. 많은 항암제들이 DNA 복제를 방해함으로써 작용하기 때문에, 우리는 약물 치료가 화학 요법 후 암 진화에 영향을 미칠 수 있는 p53-proficient세포에서도 WGD를 촉진할 수 있다고 추측한다.
replicative stress는 장기간의 checkpoint-dependent G2 arrest 후 mitotic bypass를 유도하며, ATR은 DNA 손상 신호에 주로 책임이 있다. 이 mitotic bypass는 일반적으로 CDK 활성에 의해 억제되는 APC/C, APC/CCdh1의 G1 형태의 활성화를 필요로 한다. 인간 세포의 DNA damage checkpoint는 mitotic CDK의 Wee1 dependent inhibition을 통한 mitosis로의 진입을 차단한다. 이러한 CDK의 억제는 mitosis 진입을 방지하기에 충분하며 mitotic bypass에 필수적이지만 APC/CCdh1을 활성화하기에는 충분하지 않다. 대신, mitotic bypass는 추가적인 CDK inhibitor, p21을 필요로 하며, 이 경우, Wee1과 평행한 경로에서 p53을 통해 그 축적 또한 체크포인트 활성화에 의존한다. G2(높은 mVenus-Gem)에서 G1(낮은 mVenus-Gem)로 전환하는 데 걸리는 시간은 우리 실험에서 매우 다양했으며, 이는 APC/C의 스위치와 같은 활성화에 관련된 피드백 루프의 일부 또는 전부가 정상적인 세포 주기에서 anaphase 전환으로 완전히 작동하지 않음을 시사한다.
지속적인 cyclin E 발현 후 보이는 mitotic bypass는 aphidicolin에 의해 유도된 bypass와 유사한 속도를 가지며, cyclin E-CDK2의 진동이 endoreduplication에 필수적이지 않음을 나타낸다. 이것은 초파리에서 자연적으로 발생하는 endoreduplication 사이클과 대조되는데, 초파리는 origin licensing를 촉진하기 위해 낮은 cyclin E 기간을 필요로 하고 복제를 촉진하기 위해 높은 cyclin E 기간을 필요로 한다. 사람의 세포에서 cyclin E 발현이 직접적으로 라이센싱을 억제하지는 않는 것으로 보인다. 예를 들어, U2OS 셀에서 cyclin E의 과발현은 G1 단계 동안 MCM 로딩 속도에 영향을 미치지 않는다(그림 S2B 및 S2D). 더욱이, cyclin E는 Cdc6.37의 APC/C-dependent degradation을 방지함으로써 라이센싱에 긍정적인 역할을 한다. 이들 셀이 S phase에 진입할 때 나타나는 MCM 로딩 감소는 cyclin E 발현이 G1 phase를 단축시켜 라이센싱에 사용 가능한 시간을 단축시키기 때문이다(그림 S2B 및 S2C). 이러한 결과는 또한 cyclin E-CDK2 phosphorylation가 아닌 인간 cyclin A-CDK2가 시험관 내 APC/CCdh1 활성을 억제할 수 있다는 것을 보여주는 생화학 실험과 일치한다. 따라서, 우리의 결과는 cyclin E 과발현이 replication origin licensing이나 APC/CCdh1 활성을 직접적으로 억제하지 않는다는 생각과 일치한다.
aphidicolin에 의해 유도된 mitotic bypass 후, p53-proficient RPE1 세포는 노화와 같은 상태에서 정지한다. 이것은 G2에서 p53의 일시적인 유도가 mitotic bypass 후 노화 진입을 촉발한다는 것을 보여주는 이전 연구와 매우 관련이 있다. cyclin E 발현은 이러한 노화 진입을 방지하고 이러한 노화 세포가 endoreduplication을 완료하도록 할 수 있다. 예를 들어, CCNE1을 증폭함으로써, 노화에 들어가지 않고 endoreduplicate를 위해 준비되어야 한다. driver acquisition 전에 mitotic bypass및 노화가 발생하는 다른 경우, 후속 E2F deregulation 또는 p53 손실은 세포를 tetraploid 세포로서 노화로부터 다시 순환으로 몰아갈 수 있다.
이전 연구에 따르면 DSB는 p53이 부족한 세포에서 WGD를 구동할 수 있는 반면, 우리의 결과는 replicative stress에 의해 구동되는 WGD는 p53을 필요로 한다는 것을 보여준다. 두 경우 모두 WGD의 기본 메커니즘은 동일하며, 확장된 checkpoint-dependent CDK 불활성화는 APC/CCdh1 활성화 및 후속 mitotic bypass를 가능하게 한다. 그러나 DSB 구동 메커니즘은 DSB가 WGD를 방지하는 p53-dependent G1-arrest를 유발하기 때문에 p53 결핍을 필요로 한다. 여기서 우리가 replicative stress에 대해 설명하는 메커니즘은 p21로부터의 추가적인 CDK 억제가 mitotic bypass에 필수적이기 때문에 p53 숙련도를 필요로 한다; replicative stress가 p53-dependent G1-arrest를 유도하지 않기 때문에 p53 손실은 필요하지 않다. DSB 구동 메커니즘은 체크포인트 신호를 생성하기 위해 텔로미어 소모를 필요로 하며 p53 비활성화를 필요로 한다. 이러한 유전적 사건들은 모두 암에서 흔하며 따라서 두 경로 모두 암에서 중요한 역할을 할 수 있다.
우리의 결과는 viral oncogenes가 항상 p53을 완전히 비활성화시키지 않을 수 있다는 것을 보여준다. 예를 들어, SV40 large T antigen에 의해 변형된 BJ 세포는 zeocin 또는 aphidicolin으로 처리될 때 여전히 p21을 발현할 수 있다(그림 5H). 또한 DSB 구동 및 replicative stress 구동 메커니즘 모두 p53 기능을 저하시킨 BJ-LT와 같은 세포에서 작동할 수 있다. 일반적인 p53 돌연변이의 능력을 DSB 유도와 replicative stress-driven endoreduplication 모두를 촉진하는 것은 흥미로울 것이다. p53은 고전적으로 tumor suppressor gene로 간주된다. 그러나 p53 null 돌연변이는 암에서 드물고, 암에서 p53 돌연변이의 분포에 대한 연구는 더 미묘한 시각을 가져왔다 우리의 결과는 p53의 이러한 관점에 들어맞고 p53이 replicative stress 중심의 WGD를 촉진함으로써 실제로 암 진화에 기여할 수 있음을 제안한다.