
서카디언 리듬에 따른 종양 침윤 및 CD8+ T 세포의 기능이 면역요법의 효능을 결정한다
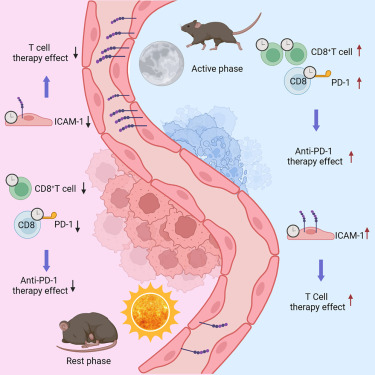

Abstract
종양 내 침윤 림프구, 특히 CD8+ T 세포의 질과 양은 종양 성장 제어와 면역요법 반응에 중요한 요소입니다. 여기에서 우리는 쥐와 인간 암에서 이러한 요소들이 서카디언 리듬(생체 시계)에 따라 변동한다는 것을 보여줍니다. 이 변동은 백혈구의 내인성 서카디언 시계와 종양 미세환경의 내피 세포 서카디언 시계에 의해 조절되는 리드미컬한 백혈구 침윤에 의해 주도됩니다. 이러한 리듬을 치료적으로 활용하기 위해, 우리는 키메라 항원 수용체 T 세포 요법과 면역 체크포인트 억제제의 효능이 하루 중 치료 시간을 조정함으로써 향상될 수 있음을 입증했습니다. 더욱이, 쥐 종양 모델에서 시간에 따라 달라지는 T 세포 서명이 흑색종 환자의 전체 생존율을 예측하고, 항-PD-1 요법에 대한 반응과 상관관계가 있음을 발견했습니다. 우리의 데이터는 종양 미세환경에서 서카디언 리듬의 기능적 중요성을 보여주며, 이러한 특징을 활용하여 미래 임상 시험 설계 및 환자 치료를 개선하는 것이 중요함을 시사합니다.
Figures
Leukocyte infiltration of tumors exhibits circadian oscillations
– 종양으로의 백혈구 침윤이 서카디언 리듬을 보인다
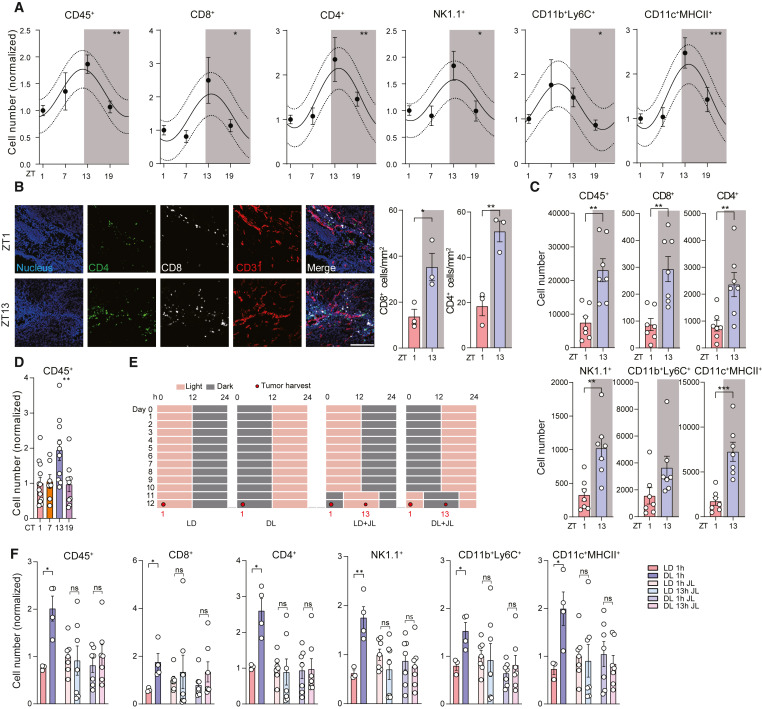

Fig. 1. 수확 시간대가 종양 침윤 백혈구 수에 미치는 영향
(A) 이 그림은 B16-F10-OVA 종양에서 하루 중 4개의 다른 시간대에 수확한 종양 침윤 백혈구(TILs)의 총 수를 보여줍니다. 결과는 저녁(ZT13)에 백혈구 수가 최고조에 달했음을 나타냅니다.
(B) 이 그림은 ZT1(아침)과 ZT13(저녁)에서 수확한 B16-F10-OVA 종양에서 CD4+와 CD8+ T 세포의 수를 비교한 것입니다. 결과는 저녁에 더 많은 T 세포가 존재함을 보여줍니다.
(C) 이 그림은 자발적 흑색종 모델(Tyr::CreERT2; PtenloxP; BRafCA)에서 ZT1(아침)과 ZT13(저녁)에서 수확한 종양 침윤 백혈구 수를 보여줍니다. 저녁에 백혈구 수가 더 많음을 확인합니다.
(D) 이 그림은 쥐를 지속적인 어둠 조건에 두었을 때도 시간대에 따라 종양 침윤 백혈구 수가 변함을 보여줍니다. 이는 이러한 변동이 단순히 빛-어둠 주기에 대한 반응이 아니라 진정한 서카디언 리듬임을 나타냅니다.
(E) 이 그림은 쥐를 정상적인 빛-어둠 주기에서 역전된 어둠-빛 주기로 전환했을 때 종양 침윤 림프구 수가 역전됨을 보여줍니다. 이는 빛이 직접적인 원인은 아니지만 새로운 조명 환경에 맞추어 조정될 수 있음을 나타냅니다.
(F) 이 그림은 쥐를 급성 시차 프로토콜에 따라 6시간 지연시킨 후, 이전에 평가된 아침과 저녁 시간대에 조직을 수확했을 때 시간대에 따른 TIL 수 차이가 사라졌음을 보여줍니다. 이는 서카디언 리듬이 빛에 의해 조정될 수 있음을 나타냅니다.
[Fig 1A] 하루 중 4개의 다른 시간대(zeitgeber time [ZT])에 수확한 B16-F10-OVA 종양에서 정상화된 총 종양 침윤 백혈구 수; n = 15, 11, 11, 12 마리의 쥐, 4개의 독립 실험에서 얻은 데이터, 코시너 분석. 음영 부분은 어두운 단계를 나타냅니다.
[Fig 1B] ZT1 또는 ZT13에서 수확한 B16-F10-OVA 종양에서 CD4+ 및 CD8+ T 세포의 이미징(왼쪽)과 정량화(오른쪽); n = 3 마리의 쥐, 독립형 Student’s t-검정, 스케일 바: 200 μm.
[Fig 1C] ZT1 또는 ZT13에서 수확한 Tyr::CreERT2, BRafCA, PtenloxP 쥐에서 종양 침윤 백혈구 수; n = 7 마리의 쥐, 독립형 Student’s t-검정.
[Fig 1D] 지속적인 어둠 조건(서카디언 시간 [CT])에서 하루 중 다른 시간대에 수확한 B16-F10-OVA 종양에서 정상화된 총 종양 침윤 백혈구 수; n = 12, 7, 10, 9 마리의 쥐, 3개의 독립 실험에서 얻은 데이터, 일원배치 분산분석(one-way ANOVA).
[Fig 1E] 빛:어둠(LD), 역전 어둠:빛(DL), 시차(JL) 조건의 빛 일정. 음영 부분은 어두운 단계를 나타내며, 숫자는 수확 시간을 나타냅니다.ains에 대한 motifs가 상당히 풍부한 Transcription factors (grouped by family).
[Fig 1F] 빛:어둠(LD, n = 3 마리의 쥐), 역전 어둠:빛(DL, n = 4 마리의 쥐), 또는 해당 시차(JL, n = 7 마리의 쥐) 조건에서 주기 시작 후 지정된 시간 지점(1 또는 13 시간)에 수확한 B16-F10-OVA 종양에서 종양 침윤 백혈구 수, 3개의 독립 실험에서 얻은 데이터, 독립형 Student’s t-검정. 음영 부분은 어두운 단계를 나타냅니다. 모든 데이터는 평균 ± SEM으로 표현되며, ns는 유의미하지 않음을, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001을 나타냅니다. Figure S1도 참조하십시오.
Endothelial cells gate circadian leukocyte infiltration
– 혈관내피세포는 하루 리듬에 따른 백혈구 침투를 조절합니다.
Fig. 2. 비정상적인 CG DNA methylation은 primed reprogramming 13일 이후에 획득되며 naive-hiPS 세포에는 존재하지 않습니다.
(A) LFA-1 인테그린을 차단하는 항체(anti-αLβ2)를 사용하여 24시간 동안 급성으로 백혈구 침투를 차단한 결과, 저녁(ZT13)에 종양 내 백혈구 수가 크게 감소하고 시간에 따른 차이가 사라졌습니다. 이는 백혈구가 혈액에서 종양으로 들어오는 것이 시간에 따른 차이를 만드는 데 중요한 역할을 한다는 것을 의미합니다.
(B) 종양으로의 백혈구 이동은 시간에 따라 크게 달라졌으며, 저녁에 더 많은 백혈구가 종양에 도달했습니다. 이는 앞서 언급한 침투 차단 데이터와 일치합니다.
(C) 세포 간 접착 분자 1(ICAM-1)은 저녁(ZT13)에 내피 세포에 의해 훨씬 더 많이 발현되었으며, 반면에 혈관 세포 접착 분자 1(VCAM-1)과 E-셀렉틴은 시간에 따른 차이를 보이지 않았습니다. 이러한 현상은 면역 결핍이 높은 NSG 마우스에서도 관찰되었으며, 이는 이러한 차이가 종양 백혈구 침투의 진동에 의해 발생한 것이 아님을 나타냅니다.
(D) Bmal1ΔEC 마우스에서는 종양 내피 세포에서 ICAM-1 발현의 시간 차이가 완전히 사라졌지만, 혈관 밀도는 야생형 마우스와 유사했습니다.
(E) Bmal1ΔEC 마우스에서 총 종양 백혈구 수와 체외에서 이식된 백혈구의 종양 침투는 더 이상 시간에 따라 달라지지 않았습니다.

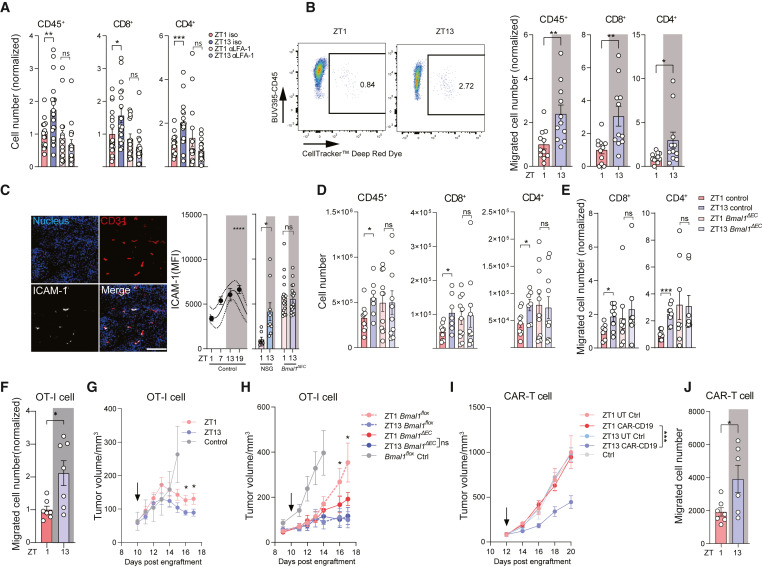
[Fig 2A] LFA-1 인테그린을 차단하는 항체(anti-αLβ2)를 사용하여 24시간 동안 급성으로 백혈구 침투를 차단한 결과, 저녁(ZT13)에 종양 내 백혈구 수가 크게 감소하고 시간에 따른 차이가 사라졌습니다. 이는 백혈구가 혈액에서 종양으로 들어오는 것이 시간에 따른 차이를 만드는 데 중요한 역할을 한다는 것을 의미합니다.
[Fig 2B] primed-hiPS 세포에서 CG-DMR (differentially methylated regions)의 60.4%가 hES 세포에 비해 hypo-methylated 되어 있었고 fibroblasts에 비해 methylation levels에서 20% 미만의 차이를 보여 somatic cell epigenetic memory를 나타냈으며, hES 세포에 비해 hypo-methylated된 CG-DMR의 24.2%가 추가로 primed-hiPS 세포에서 fibroblasts에 비해 더 높은 methylation을 보여 partial epigenetic memory를 나타냄.
반대로, hyper-methylated CG-DMR의 대부분(54.2%)은 reprogramming 중에 획득한 비정상적인 DNA methylation을 보였으며, methylation levels이 fibroblasts와 hES 세포 모두보다 20% 이상 높았음.
[Fig 2C] Time-course analysis 결과, primed reprogramming 13~21일 사이에 비정상적인 methylation이 나타나기 시작하여 21일과 3~10일 사이에 계속 증가하는 것으로 나타남.
[Fig 2D] memory CG-DMR의 경우, primed reprogramming에서 minor transient demethylation (mCG/CG <0.1) 발생했으며, 이는 global CG methylation change (Fig.1b)와 일치.
[Fig 2D,E] 세포를 naive medium으로 전환하면 13일째에 memory CG-DMR에서 상당한 demethylation이 발생.
- 비정상적인 CG methylation이 early reprogramming동안 OKSM induction 시 축적되지 않으며, primed reprogramming 13일 이후에야 나타나기 시작함.
- primed-hiPS 세포의 비정상적인 CG hyper-methylation loci는 naive reprogramming에서는 비정상적이지 않음. 이는 비정상적인 hyper-methylation이 naive reprogramming이 아닌 primed reprogramming의 특징이라는 것을 나타냄.
[Fig 2E] 알려진 ICR (imprint control regions)에서 CG methylation을 분석한 결과, imprint는 7~13일 사이에 CG methylation을 잃기 시작하며, allele-specific methylation의 full loss는 naive reprogramming 21일 이후까지 발생하지 않는 것으로 나타남.
- imprinted loci의 demethylation이 naive conditions에서 세포가 더 오래 배양할수록 더 광범위해지며, imprints가 naive reprogramming 13 일째에도 유지될 수 있음을 시사.
TNT reprogramming resets the epigenome
– early development 과정에서 pre-implantation embryo는 global demethylation wave와 관련된 epigenetic reset을 겪으며, 이 과정에서 genomic imprints가 demethylation으로부터 보호됨.
– embryonic development 중에 관찰되는 demethylation과 유사한 transient naive-like state를 통해 reprogramming함으로써 somatic cell epigenetic memory와 비정상적인 DNA methylation을 피할 수 있다는 가설 세움. 따라서 두 가지 실험 시스템 계획.
- fibroblasts medium에서 초기 7일간 배양한 후 5일간 transient naive culture treatment로 fibroblasts를 reprogramming한 다음 나머지 reprogramming을 위해 primed medium에서 배양하여 transient-naive-treatment hiPS 세포 (TNT-hiPS 세포)를 생성 (Fig.3a).
- 먼저 naive culturing을 연장하여 naive-hiPS cell colonies를 확립한 다음, 세포를 primed pluripotent state로 전환하여 naive-to-primed hiPS 세포 (NTP-hiPS 세포)를 생성 (Fig.3a).
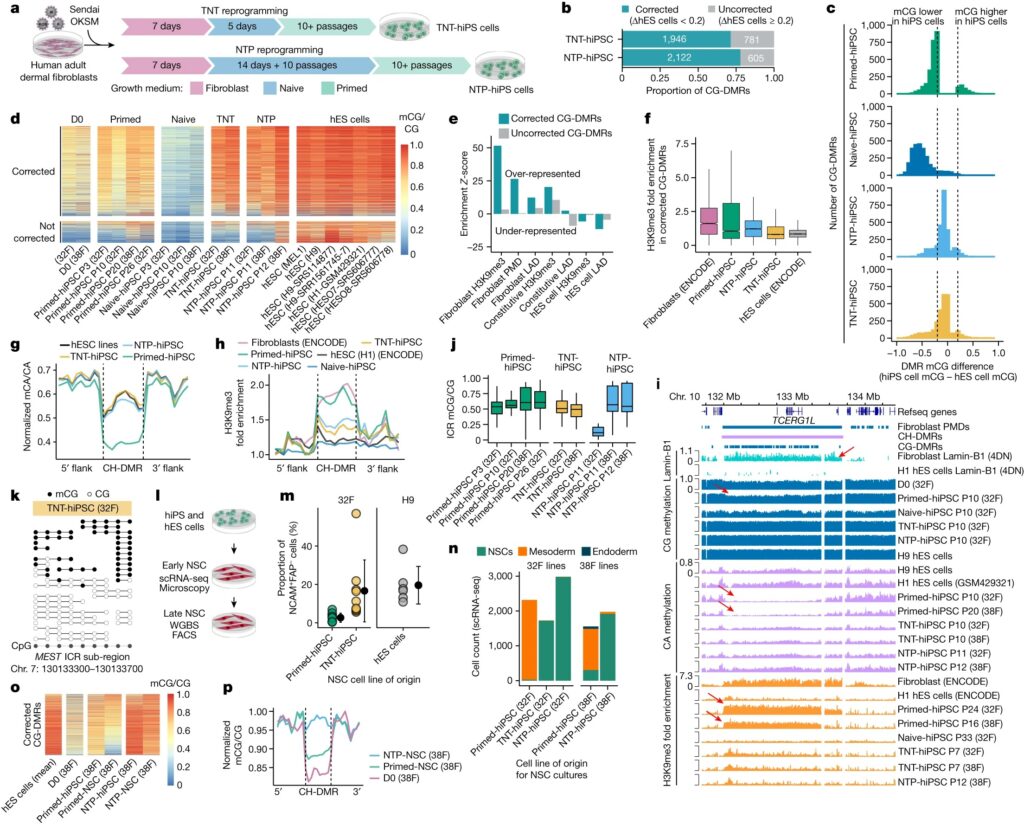

Fig. 3. naive state를 통한 reprogramming은 somatic cell memory를 지우고 hES 세포와 매우 유사한 hiPS 세포를 생성합니다.
(A) TNT-hiPS 및 NTP-hiPS 세포에 대한 새로운 reprogramming 전략.
(B) TNT 및 NTP reprogramming에 의해 0.2mCG/CG 미만의 차이로 corrected primed-hiPS 세포 및 hES 세포에 대한 CG-DMR의 비율.
(C) CG-DMR에서 hiPS와 hES 세포 간의 DNA methylation 차이.
(D) corrected (위쪽) 및 uncorrected (아래쪽) CG-DMR의 methylation levels.
(E) repressive chromatin에서 corrected 및 uncorrected CG-DMR의 Enrichment permutation testing.
(F) corrected CG-DMR에서 H3K9me3 enrichment.
(G) hypo-methylated CH-DMR에서 CA methylation의 Aggregate profile.
(H) hypo-methylated CH-DMR에서 H3K9me3 enrichment.
(I) PMD, fibroblast LAD 및 CG-DMR cluster를 교차하는 CH-DMR의 Genome track.
(J) ICR의 CG methylation.
(K) MEST ICR에서 읽은 WGBS.
[Fig. 3B-D] primed-hiPS cell과 hES cell lines 사이에서 검출된 CG-DMR을 평가할 때, 대부분의 CG-DMR이 TNT-hiPS(71.3%) 및 NTP-hiPS(77.8%) 세포 모두에서 hES 세포와 매우 유사한 상태로 epigenetic correction을 보이는 것을 관찰
[Fig. 3E] corrected CG-DMR은 fibroblast에서 repressive histone modification H3K9me3를 특징으로 하는 영역에서 매우 풍부하지만(z-score = 38.9; FDR <0.01), hES cell-specific H3K9me3 영역에서는 고갈된 것을 확인(z-score = -4.5; FDR <0.01).
일관되게, corrected CG-DMR은 fibroblast의 partially methylated domains (PMD)(z-score = 25.8; FDR <0.01)과 lamina associated domains (LAD)(z-score = 10.6; FDR <0.01)에서 과도하게 나타났는데, 이는 gene-poor, repressive하며 higher order genome architecture와 관련된 heterochromatin의 큰 도메인에서 H3K9me3와 함께 발생하는 것으로 알려져 있음.
[Fig. 3F] corrected CG-DMR과 교차하는 fibroblast에서 H3K9me3가 enriched된 영역은 hES 세포와 더 유사한 TNT-hiPS 및 NTP-hiPS 세포에 비해 primed-hiPS 세포에서 더 높은 H3K9me3를 보임
- 이는 epigenetic memory를 특징으로 하는 repressive chromatin domains이 TNT reprogramming에 의해 재설정됨을 시사.
[Fig. 3G] hypo-methylated CH-DMR에서 CA methylation을 검사한 결과, TNT-hiPS 및 NTP-hiPS 세포는 hES 세포와 매우 유사한 CA methylation profile을 가지고 있으며, 이는 hyper-methylated CH-DMR과 달리 primed-hiPS 세포에서 관찰된 낮은 CA methylation level과는 구별됨.
[Fig. 3H] 우리는 primed-hiPS 세포의 hypo-methylated CH-DMR에서 fibroblasts와 유사한 수준으로 강력한 H3K9me3 enrichment를 관찰했지만, TNT-hiPS 및 NTP-hiPS 세포는 hES 세포와 더 유사했으며 H3K9me3가 현저히 적음.
[Fig. 3I] Fibroblasts의 10번 chromosome에서는 lamin-B1에 대해 농축되었지만 hES 세포에서는 농축되지 않은 1.7-Mb CH-DMR을 발견했으며, 이는 175개의 작은 CG-DMR cluster에 걸쳐 있고 더 큰 fibroblast PMD와 교차하며 fibroblast와 primed-hiPS 세포에서는 H3K9me3의 5배 이상의 농축을 보이지만 TNT-hiPS 및 NTP-hiPS 세포에서는 그렇지 않은 것으로 나타남.
- nuclear lamina와 관련된 큰 repressive chromatin 도메인이 primed-hiPS 세포에서 epigenetic memory를 보유하고 있음을 나타냄.
- TNT-hiPS 및 NTP-hiPS 세포에서 CG 및 CH methylation 및 H3K9me3의 correction은 hiPS 세포의 epigenetic memory의 대부분을 corrected 할 수 있음을 보여주며, TNT reprogramming이 기존의 reprogramming에서 달성되는 것 이상으로 chromatin architecture를 재구성한다는 것을 시사.
[Fig. 3J] 이전 연구에 따르면 naive culturing은 genomic imprinting의 손실을 유발하며, 이는 re-priming해도 복구되지 않음. 이와 대조적으로, TNT-hiPS 세포가 imprinting을 나타내는 CG methylation patterns을 가지고 있음을 관찰.
[Fig. 3K] single DNA molecules을 대표하는 WGBS reads를 분석한 결과, fibroblast와 유사하게 TNT-hiPS 세포의 ICR에서 unmethylated and methylated molecules의 비율이 동일한 것으로 나타남
[Fig. 3J] 이는 imprinted loci에서 methylation levels의 편차가 증가하는 것을 관찰한 NTP-hiPS 세포와는 대조적임.
- 이는 TNT-hiPS 및 NTP-hiPS 세포가 적절한 X chromosome inactivation을 특징으로 한다는 것을 나타냄.
Correction persists through differentiation
이전 연구에 따르면 primed-hiPS 세포의 epigenetic memory와 aberrations은 differentiation 후에도 지속될 수 있으며, 이는 resulting cells에 기능적으로 영향을 미칠 수 있음.
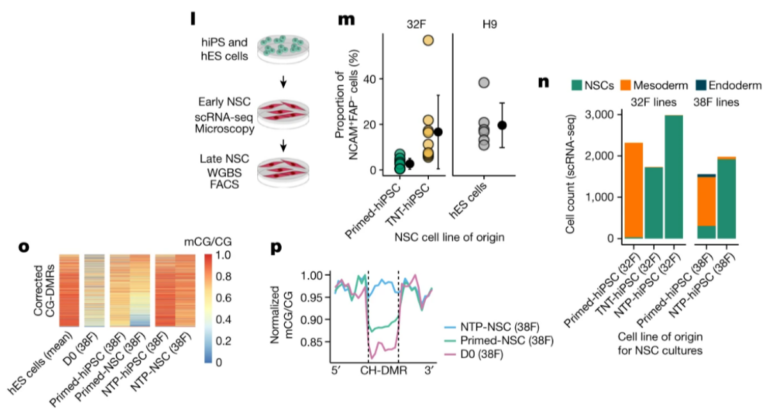

Fig. 3. naive state를 통한 reprogramming은 somatic cell memory를 지우고 hES 세포와 매우 유사한 hiPS 세포를 생성합니다.
(L) NSC differentiation 및 프로파일링의 개략도.
(M) NSC로 differentiation되는 동안 NCAM+FAP- 세포의 비율.
(N) single-cell RNA-seq (scRNA-seq)에 의해 early NSC cultures에서 검출된 다양한 세포 유형의 비율.
(O) hiPS 세포 및 derived NSC cultures에서 NTP reprogramming(as in Fig. 3d)에 의해 corrected CG-DMR의 Methylation levels.
(P) NSC 및 progenitor fibroblasts의 hypo-methylated CH-DMR에서 확인된 CG methylation (flank-normalized mCG/CG).
* NCAM : neural marker, FAP : fibroblast marker
[Fig. 3L] primed-hiPS, TNT-hiPS 및 NTP-hiPS 세포를 neural stem cells (NSC)로 differentiation시켜 CG-DMR correction이 유지되는지 여부를 테스트.
[Fig. 3M] differentiating culture에서 NCAM+FAP- 세포의 FACS 정량화 결과, TNT-hiPS 세포가 hES 세포와 유사한 속도로 NSC로 더 효율적으로 differentiation됨.
[Fig. 3N] 7%의 mesoderm-like cells (마커 BMP4, HAND1 및 TGFB1에 의해 정의됨)로 구성되어 있으며, 이는 ibroblast-derived TNT-hiPS 세포 (0.35%) 및 NTP-hiPS 세포(0.06-0.27%)에서 생성된 NSC cultures에는 존재하지 않음.
[Fig. 3O] primed-hiPS cell derived NSCs의 CG-DMR에서 hypo-methylation이 지속되는 반면, NTP-hiPS cell derived NSC의 경우 epigenetic correction이 유지.
[Fig. 3P] primed-hiPS cell derived NSCs는 실제로 partial CG methylation을 유지했으며, 이는 repressive chromatin의 리모델링을 시사하는 높은 CG methylation levels을 보인 NTP-hiPS 세포와는 대조적.
- 이러한 결과는 primed-hiPS 세포의 epigenetic memory가 differentiation efficiency를 손상시키고 differentiation 내내 지속된다는 것을 나타냄.
Isogenic evaluation of hiPS and hES cells
일련의 isogenic reprogramming experiments를 설계하여 hiPS 세포와 hES 세포를 명확하게 비교.
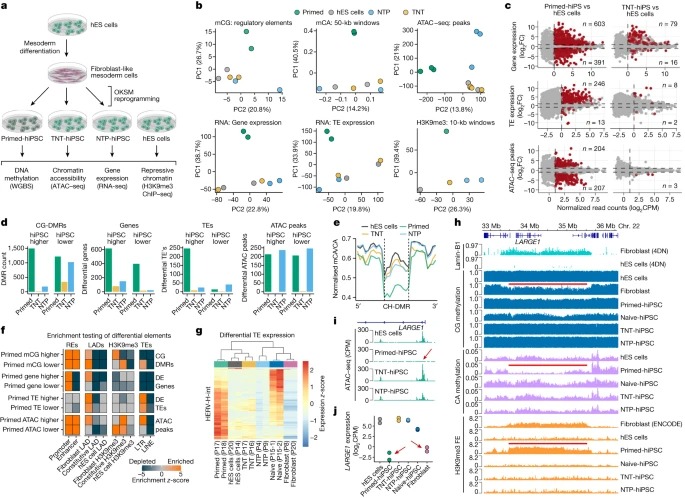

Fig. 4. isogenic differentiation 및 reprogramming system은 TNT reprogramming이 epigenome resetting을 향상시킨다는 것을 확인합니다.
(A) hES 세포를 fibroblast-like cells로 differentiation시킨 다음 primed, TNT 및 NTP 방법을 사용하여 hiPS 세포로 reprogramming하는 실험 설계.
(B) GeneHancer elements의 CG methylation, 50-kb genome windows의 mCA/CA, 피크의 normalized ATAC–seq read counts, normalized global gene expression, normalized global transposable element (TE) expression 및 normalized H3K9me3 ChIP–seq read counts에 대한 Principal component analysis.
(C) hiPS 세포와 hES 세포의 유전자 발현(RNA-seq에 의해 결정됨), TE 발현(RNA-seq), chromatin accessibility (ATAC-seq)에 대한 Differential-testing MA plots.
(D) CG-DMR, 유전자 발현, TE 발현 및 ATAC-seq 피크에 대한 hES 세포와 hiPS 세포 유형의 Differential testing.
(E) hypo-methylated CH-DMR에서 CA methylation levels의 Aggregate profile plot.
(F) differential elements의 Permutation testing enrichment (z-scores).
(G) hES 세포와 primed-hiPS 세포 간에 differentially expressed되는 HERV-H-int elements의 Relative expression heatmap.
(H) hES 세포와 primed-hiPS 세포에서 검출된 CH-DMR 영역의 Genome track 및 관련 epigenomic features.
(I) LARGE1 promoter에서 Normalized ATAC–seq signal.
(J) isogenic hES 세포, hiPS 세포 및 progenitor fibroblasts에서 LARGE1의 유전자 발현.
[Fig 4A] primed-hiPS, TNT-hiPS 및 NTP-hiPS cell protocols을 사용하여 이러한 secondary fibroblasts를 reprogramming하고 WGBS, RNA-seq, transposase-accessible chromatin에 대한 sequencing (ATAC-seq) 및 H3K9me3 ChIP-seq을 수행
[Fig 4B] genetic differences를 통제하더라도 TNT-hiPS 세포는 hES 세포와 일관되게 매우 유사한 반면, primed-hiPS 세포는 molecularly하게 구별된다는 것을 확인.
[Fig 4C,D] isogenic primed-hiPS 세포와 hES 세포 사이에서 differentially expressed되는 994개의 유전자를 확인했지만, 이러한 차이는 TNT-hiPS와 NTP-hiPS 세포에서 크게 개선되어 각각 95개와 165개의 유전자만 differentially expressed됨.
chromatin accessibility의 차이를 테스트했을 때, hES 세포와 primed-hiPS 세포 간에는 411개의 ATAC-seq 피크가 다르게 관찰된 반면, hES 세포와 TNT-hiPS 세포 간에는 3개의 피크만 다르게 관찰되어 사실상 구별할 수 없었음. NTP-hiPS 세포는 483개의 differential peaks를 보였지만 primed-hiPS 세포와 같은 방향은 아니었음.
primed-hiPS 세포에서 246개의 up-regulated와 13개의 down-regulated transposable elements를 확인함. 특히, 이러한 차이는 TNT reprogramming에 의해 거의 완전히 사라져 8개의 up-regulated와 2개의 down-regulated transposable elements만 남았지만, NTP-hiPS 세포는 여전히 65개의 differentially expressed transposable elements를 보임.
primed-hiPS 세포의 경우 2,709개의 CG-DMR을 검출했고, TNT-hiPS 세포의 경우 358개, NTP-hiPS 세포의 경우 1,200개만 검출.
[Fig 4E] TNT-hiPS 및 NTP-hiPS 세포는 primed-hES 세포와 달리 CH-DMR에서 origin hES 세포와 유사한 CA methylation levels를 보임
[Fig 4F] long terminal repeat (LTR) transposable elements에서 primed-hiPS 세포 ATAC-seq 피크의 농축이 reduced CG methylation와 함께 발생하는 것을 관찰.
[Fig 4G] primed-hiPS 세포에서 up-regulated transposable elements는 주로 human endogenous retrovirus subfamily H (HERV-H) elements (80%, 246개 중 197개)와 그 flanking LTR7 서열이며 primed-hiPS 세포는 naive-hiPS 세포에서 발현되는 elements와 비교해 이들 elements의 뚜렷한 copies를 발현.
[Fig 4F] differential DNA methylation, gene expression 및 chromatin states 간의 관계를 분석할 때, 우리는 primed-hiPS 세포에서 유의하게 다른 것으로 확인된 elements에 대해fibroblast-associated repressive chromatin 도메인이 매우 풍부하다는 것을 관찰.
[Fig 4H] 22번 chromosome에서 약 2Mb의 fibroblast LAD를 검사했을 때, primed-hiPS 세포는 fibroblast와 유사하게 H3K9me3 농축이 수반된 PMD를 가지고 있지만 isogenic TNT-hiPS 세포, NTP-hiPS 세포 및 hES 세포와는 구별되는 것을 관찰.
[Fig 4I,J] 이 fibroblast LAD 내에서 LARGE1 promoter는 강력한 transcriptional repression과 함께 primed-hiPS 세포에서 chromatin accessibility를 보이지 않음.
Improved differentiation of TNT-hiPS cells
– iPS 세포의 epigenetic memory가 differentiation에 영향을 미친다는 상당한 증거가 있지만, iPS 세포와 ES 세포 간의 기능적 차이는 여전히 논쟁의 여지가 있음.
– primary human dermal fibroblasts (HDF), keratinocytes (NHEK 세포), mesenchymal stem cells (MSC) 및 hES cell-derived isogenic secondary fibroblasts에서 reprogramming된 독립적인 hiPS cell lines을 추가로 생성하여 primed 및 TNT-hiPS 세포 differentiation capacity의 차이를 종합적으로 테스트(Fig. 5a)
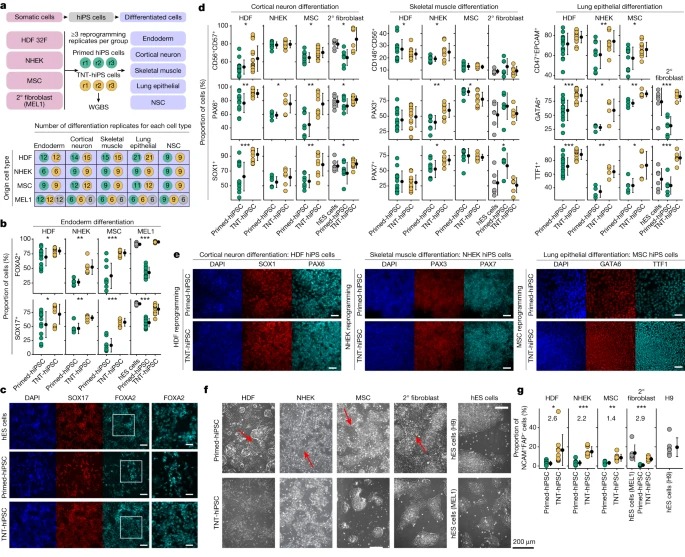

Fig. 5. Multi-lineage reprogramming 및 differentiation은 TNT reprogramming이 differentiation을 향상시킨다는 것을 확인합니다.
(A) multi-lineage primed 및 TNT reprogramming 및 5가지 세포 유형으로의 differentiation을 위한 실험 설계.
(B) secondary fibroblasts에서 유래한 hiPS 세포에 대한 Endoderm differentiation quantification, immunofluorescence analysis에 의해 FOXA2 및 SOX17에 positive인 세포의 비율.
(C) secondary fibroblasts에서 유래한 hiPS 세포의 endoderm differentiation에서 FOXA2 및 SOX17의 immunofluorescence analysis Representative images.
(D) cortical neuron differentiation을 위한 CD56, CD57 (FACS), PAX6 및 SOX1 (immunofluorescence), skeletal muscle differentiation을 위한 CD146, CD56(FACS), PAX3 및 PAX7(immunofluorescence), lung epithelial differentiation를 위한 CD47, EPCAM(FACS), GATA6 및 TTF1(immunofluorescence)을 사용한 immunofluorescence와 FACS로 정량화한 hiPS cell lines의 multi-lineage cell differentiation.
(E) cortical neurons의 경우 SOX1 및 PAX6, skeletal muscle의 경우 PAX3 및 PAX7, lung epithelial cells의 경우 GATA6 및 TTF1을 사용한 cell differentiation에 대한 immunofluorescence analysis의 Representative images.
(F) NSC로 differentiation되는 동안 plated embryoid bodies를 passaging한 후 4일 뒤에 촬영한 Phase-contrast images.
(G) NSC differentiation 중 embryoid bodies plating 후 NCAM+FAP- 세포의 백분율(FACS analysis 결과).
[Fig 5A] FACS 및 immunofluorescence 정량화를 통해 모든 hiPS cell lines의 differentiation capacity를 광범위하게 테스트함.
[Fig 5B,C] definitive endoderm differentiation를 시킬 때, 우리는 origin cell type에 관계없이 TNT-hiPS 세포가 primed-hiPS 세포에 비해 definitive endoderm으로 differentiation하는 데 일관되게 더 효율적이라는 것을 관찰함.
[Fig 5D,E] hES 세포, primary HDF 및 MSC에서 유래한 secondary fibroblasts에서 생성된 TNT-hiPS 세포는 primed-hiPS 세포보다 cortical neurons과 lung epithelial cells로 더 효율적으로 differentiation했으며, 이 cell types의 주요 마커를 발현하는 세포의 비율이 더 높음을 확인.
[Fig 5D,E] skeletal muscle cell differentiation의 경우, MSC, HDF 및 secondary fibroblasts에서 생성된 TNT-hiPS 및 primed-hiPS 세포 모두 유사한 효율로 differentiation됨.
[Fig 5D,E] NHEK-derived hiPS 세포의 경우, primed-hiPS 세포와 TNT-hiPS 세포 모두 비슷한 효율로 cortical neurons으로 differentiation되었지만, TNT-hiPS 세포는 primed-hiPS 세포보다 lung epithelial cells 및 skeletal muscle cell로 differentiation하는 데 더 효율적임을 확인.
[Fig 5F] NSC colonies가 형성되는 early differentiation 단계에서, 우리는 세포가 primed-hiPS 세포에서 유래했을 때 길쭉한 fibroblast-like cells의 자발적인 출현을 다시 관찰했지만, TNT-hiPS 세포에서 유래했을 때는 그렇지 않음.
[Fig 5G] NSC differentiation efficiency를 정량화한 결과, primed-hiPS 세포에서 유래한 배양액보다 TNT-hiPS 세포에서 유래한 배양액에서 NSC(NCAM+FAP-)의 비율이 일관되게 더 높았으며 hES cell lines에서 관찰된 differentiation efficiency에 더 근접한 것으로 나타남.
- 이러한 reprogramming 및 differentiation experiments는 primed-hiPS 세포의 epigenetic differences가 TNT reprogramming에 의해 약화될 수 있는 differentiation capacity 감소와 관련이 있다는 강력한 증거를 제공.
Disscussion
우리는 naive and primed reprogramming dynamics를 특성화하여 iPS 세포의 epigenetic remodelling의 본질에 대한 새로운 통찰력을 얻었으며, 이는 TNT reprogramming 전략의 개발로 이어졌습니다. 우리의 연구는 epigenetic memory가 origin 세포의 nuclear lamina와 관련된 H3K9me3로 표시된 origin 세포의 repressive chromatin 도메인에 집중되어 있음을 보여줌으로써 이전 연구를 확장합니다. 우리는 TNT reprogramming이 특히 chromatin–lamina interactions 영역에서 epigenetic memory를 효과적으로 지우고 differentiation을 개선한다는 사실을 발견했습니다. differentiation 단서에 대한 세포의 반응이 transcription factor binding에 사용할 수 있는 loci를 만들기 위해 chromatin이 공간적으로 어떻게 구성되는지에 따라 달라진다면, primed-hiPS 세포의 differentiation bias는 transcription factor binding dynamics에 영향을 미치는 heterochromatic memory에 기인할 수 있습니다.
TNT reprogramming을 통해 달성된 보다 완전한 epigenome reset은 이 전략이 human pre-implantation development 과정에서 발생하는 epigenetic reset의 측면을 모방할 수 있음을 시사합니다. 첫째, TNT reprogramming은 post-implantation lineage-specific H3K9me3가 확립되기 전에 early embryonic development 중에도 발생하는 H3K9me3 heterochromatin을 리모델링합니다. 둘째, TNT reprogramming은 pre-implantation development와 유사하게 일시적인 transient genome-wide demethylation을 촉진합니다. 셋째, genomic imprints는 pre-implantation epigenome resetting 과정에서 지워지지 않도록 보호되며, 우리의 데이터에 따르면 imprinting loss는 naive medium에서 장기간 배양할 때 나타나는 증상으로 보이기 때문에 TNT reprogramming의 일시적인 특성이 imprinting loss를 최소화할 수 있습니다.
비정상적인 HERV-H transcription이 HERV-H promoters로부터 시작된 L1 transposable element mRNA 발현의 가능성을 증가시켜 hiPS 세포에서 mutagenesis 유발로 이어지는 것으로 보고되었기 때문에, HERV-H transposable elements가 hES 세포에 비해 primed-hiPS 세포에서 더 높은 발현을 보이지만 TNT-hiPS 세포에서는 그렇지 않다는 우리의 관찰은 특히 중요합니다. 이전 연구에 따르면 hiPS 세포에 존재하는 transcriptional and epigenetic signatures는 isogenic systems에서도 donor-dependent일 수 있다고 합니다. 여기서 우리는 독립적으로 isogenic primed-hiPS 세포와 hES 세포가 유전자 발현에 상당한 차이를 보인다는 것을 확인했지만, 이러한 차이가 TNT reprogramming을 통해 제거될 수 있음을 추가로 입증했습니다. 이는 epigenome이 hES 세포와 hiPS 세포 간의 차이를 유발하는 데 중요한 역할을 한다는 것을 나타냅니다. 또한, differentiation 실험을 통해 유전적으로 일치하는 TNT-hiPS 세포가 primed-hiPS 세포보다 더 향상되고 균질한 differentiation 잠재력을 가지고 있음을 입증했습니다.
TNT reprogramming 시스템을 활용하여 epigenome을 보다 완벽하게 재설정함으로써 얻을 수 있는 기능적 이점을 밝혀냈습니다. 이 연구 이전에는 SCNT reprogramming이 DNA methylation 이상을 개선하는 유일한 방법이었습니다. 그러나 SCNT-reprogrammed 세포는 여전히 지속적인 cell-of-originH3K9me3 heterochromatin을 특징으로 할 수 있으며, 이 기술은 확장하기 어렵고 실현 불가능합니다. 우리의 연구는 TNT reprogramming이 hiPS 세포의 이러한 본질적인 특성을 극복할 수 있는 실용적이고 확장 가능한 접근법이며, 이는 이 기술의 임상 적용에 중요하다는 것을 보여줍니다. TNT reprogramming은 differentiation 개선과 함께 epigenome과 transcriptome의 high-fidelity resetting을 가능하게 하므로, 우리는 이것이 epigenetic memory와 mechanisms maintaining cell-of-origin heterochromatin을 연구하는 강력한 모델 시스템으로 간주합니다.