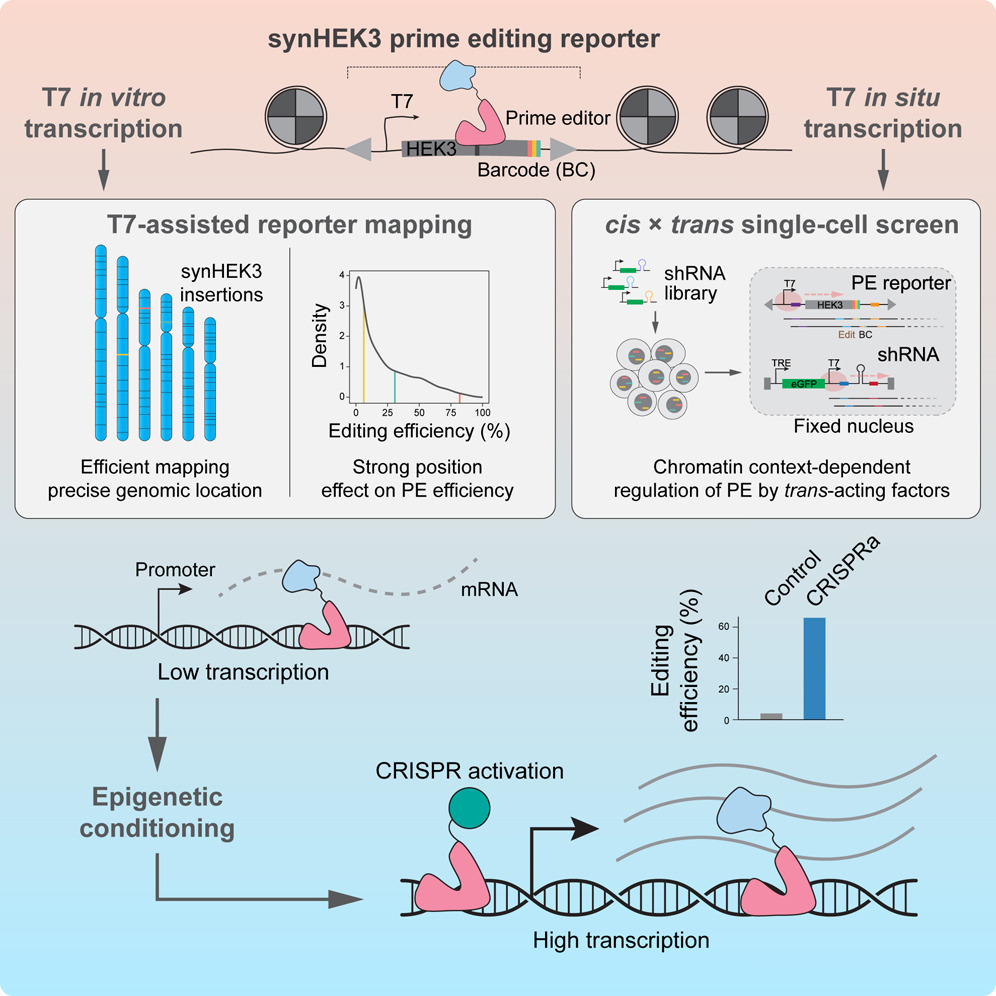
Prime editing의 염색질 맥락-의존적 통제와 후성유전학적 조절
Abstract
이 연구는 prime editing이라는 정밀한 유전체 공학 도구에 Cis-Chromatin 환경이 미치는 영향을 철저히 분석하고자 했습니다. 무작위로 통합된 리포터의 유전체 위치를 mapping하는 고감도 방법을 사용해, 동일한 target 사이트와 편집에서 편집 효율이 ∼0%에서 94%까지 크게 변동 하는 위치 효과를 발견했습니다. Prime editing 효율에 대한 위치 효과는 H3K79me2에 의해 긍정적으로, H3K9me3에 의해 부정적으로 예측되는 Chromatin 표지에 의해 잘 예측되었습니다. 이어서, Cis-Chromatin 환경에서 Trans-acting 인자가 편집 결과에 미치는 영향을 평가하기 위해 다중 교란 framework를 개발했습니다. 이 framework를 DNA 수리 인자에 적용한 결과, 상황에 따라 HLTF가 Prime editing을 억제할 수 있음을 확인했습니다. 마지막으로, 여러 증거가 활발한 전사 신장이 prime editing을 촉진함을 시사하고 있음을 보여줍니다. 이에 따라, 우리는 CRISPR를 이용한 유전자 침묵화 또는 활성화를 통해 prime editing 효율성을 각각 감소시키거나 증가 시킬 수 있음을 입증했습니다.
Figures
Design and efficient mapping of prime editing reporters
– Prime editing reporter를 설계하고 효율적인 매핑
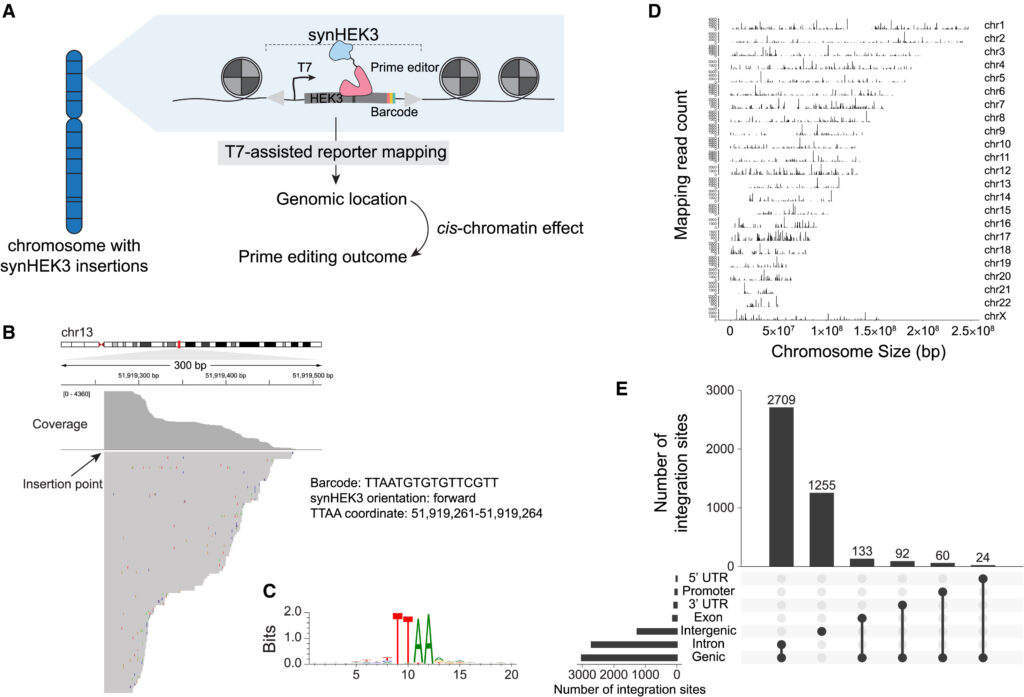
Fig. 1. SynHEK3 reporter의 통합 위치를 효율적으로 유전체 전역에서 mapping하는 방법을 개발했습니다.
(A) synHEK3 reporter 라이브러리가 유전체 내에서 무작위로 삽입되며, 각 리포터의 유전체 위치는 T7 보조 reporter mapping 방법을 사용하여 결정됩니다. Mapping된 reporter의 cis-크로마틴 맥락은 각 reporter로부터 측정 된 prime editing 결과를 모델링 하는 데 사용됩니다.
(B) 유전체 브라우저에서 독자의 정확한 좌표를 나타내는 read pileup을 보여주는 것. 바코드 서열, 방향 및 리포터의 좌표가 주석으로 달려 있습니다.
(C) synHEK3 통합 위치 주변의 20-bp 윈도우에서 모티프 enrichment 분석.
(D) 병목 현상이 일어난 풀에서 식별된 고유한 synHEK3 reportor의 커버리지 플롯 (n = 4,273). 세로 막대 길이는 읽은 수에 해당.
(E) synHEK3 통합 위치의 유전체 주석에 대한 UpSet 플롯.
[Fig 1A] cis-chromatin 효과를 검증하기 위해 synHEK3 이 통합 염색체에서 reportor의 위치를 정확히 맵핑하기 위한 실험 계획 모식도를 보여줌
[Fig 1B] synHEK3 통합의 유전체 위치를 매핑하기 위해 T7 보조 리포터 매핑을 수행. 정렬된 읽기 부분 집합을 유전체 브라우저에서 시각화 했을 때 각 부분이 날카로운 경계를 보여주며, 이는 synHEK3 리포터의 정확한 삽입 지점에 해당
[Fig 1C] 삽입 접합부의 모티프 분석 결과 PB 전이에서 예상되는 TTAA 모티프가 나타남
[Fig 1D] 이 모티프가 없는 사이트(6.4%)를 제거하고 개별 리포터의 위치로 TTAA 모티프의 유전체 좌표를 할당함. 주목할 점은 4,273개의 매핑된 사이트(42.3%)만이 고유한 바코드를 가지고 있었음.
[Fig 1E] synHEK3 리포터는 모든 주요 유전체 주석에 걸쳐 분포되어 있는 것을 보여줌
Measured the prime editing outcome by conducting transfection using pegRNA
– HEK3 타겟 서열에 CTT 삽입을 설치하기 위해 디자인된 pegRNA를 사용하여 transfection을 수행하여 prime editing결과를 측정했습니다.
Fig. 2. Chromatin 맥락은 prime editing 효율성에 주요한 영향을 미칩니다.
(A) 왼쪽: 실험의 개요도. 오른쪽: 모든 고유 바코드가 달린 synHEK3 리포터들 (n = 4,273)에서 CTT 삽입 빈도의 밀도 플롯. 빨간 선은 K562 세포의 내부 HEK3 위치에서의 prime editing 효율성 (17%)을 나타냅니다.
(B) 염색질 특성 점수에 따라 계층화 된 synHEK3 위치에서 높은 편집 가능성 (>25%) 사이트의 분율을 보여주는 히트맵. synHEK3 리포터는 크로마틴 특성 점수가 증가하는 10개의 동일한 크기의 범주로 나누어집니다. 크로마틴 특성은 prime editing 효율성과의 상관 계수 (Spearman의 r)에 따라 왼쪽에서 오른쪽으로 순서가 매겨집니다.
(C) 테스트 세트의 리포터를 사용하여 관찰된 prime editing 효율성과 예측된 효율성 사이의 산점도. 근접한 점들의 개수에 따라 점이 색으로 표시됩니다. p 값은 Spearman 상관 분석 또는 Pearson 상관 분석을 통해 결정되었습니다.
(D) 염색질 특성 점수와 prime editing 효율성 간의 Spearman의 r에 대한 산점도. 유전자 내 및 유전자 간 리포터에 대해 별도로 계산되었습니다.
(E) 유전자 근접 리포터의 prime editing 효율성. 거리는 유전자 길이에 따라 스케일링 되고 분할 되었습니다. 음의 값은 TSS 상류에 위치한 synHEK3 위치를 나타냅니다. 값이 100% 초과는 전사 종료 지점 (TTS) 하류에 위치한 synHEK3 위치를 나타냅니다. 점은 유전자의 발현 수준 (log10)에 따라 색으로 구분됩니다. TPM은 백만 개의 전사 당 발현 수를 나타냅니다.
(F) 4개의 가장 높은 편집 가능 사이트에 대한 유전체 브라우저 전개도. 통합 위치와 측정 된 편집 효율성은 윗부분의 점 플롯으로 표시되고 선택된 후성유전학적 트랙과 정렬됩니다. 각 synHEK3 삽입에 대해 편집 효율성, 편집이 있는 리드의 수 (분자), 총 리드 수 (분모)가 주석으로 달려 있습니다. 대시 세로선은 삽입 위치를 표시합니다.
(G) 유전체 내 위치에 대한 프라임 편집을 위해 디자인 된 epegRNA의 시퀀스 기반 예측 (x 축) 대 정규화 된 편집 비율 (y 축, log10 스케일)의 산점도. p 값은 Spearman 상관 분석 또는 Pearson 상관 분석을 통해 결정되었습니다.
(H) 염색질 기반 예측 (우리 모델, x 축) 대 정규화 된 편집 비율 (y 축, log10 스케일)의 산점도. p 값은 Spearman 상관 분석 또는 Pearson 상관 분석을 통해 결정되었습니다.
(I) 결합된 점수 (x 축) 대 정규화 된 편집 비율 (y 축, log10 스케일)의 산점도. p 값은 Spearman 상관 분석 또는 Pearson 상관 분석을 통해 결정되었습니다.
[Fig 2A] Prime editing result를 측정하기 위해 뭉친 세포 집단에 HEK3 타겟 서열에 CTT 삽입을 설치하도록 설계된 pegRNA를 형질 도입하였고 4일 후, 모든 고유 바코드 리포터에 대해 prime editing 효율을 측정 함.
[Fig 2B] 각 염색질 특징 점수에 따라 synHEK3 리포터를 분류했을 때, 동일한 양성화 된 맵핑 특징들이 높은 비율로 editing된 사이트와 강하게 연관되어 있음을 확인함.
[Fig 2C] 이 모델은 홀드 아웃 테스트 세트에서 prime editing 효율을 정확하게 예측하였음을 보여줌.
[Fig 2D] H3K79me2는 대부분 유전자 내부에 존재하기 때문에, 포괄적인 분석은 유전자 내부와 외부 영역에서 염색질과 prime editing 간의 상호작용 차이를 감추는 것이 가능. 따라서, 우리는 상관 분석 및 베타 회귀 모델링을 유전자 내부와 유전자 외부의 synHEK3 리포터에 대해 각각 반복 수행하여 그래프로 그린 것을 보여줌
[Fig 2E] 이 실험에서 제시한 가설은 Prime Editing이 전사적으로 활발한 영역과 활성 전사 시작점(TSS) 근처에서 가장 효율적임을 의미함. 실험 결과, Prime Editing의 효율성이 유전자의 발현 수준과 TSS에 가까운 위치와 상관관계가 있음을 확인함.
[Fig 2F] 상반되는 후생 유전학적 환경을 시각화 하기 위해, 그들은 Prime Editing 효율이 90%–94%인 상위 4개의 잘 editing되는 synHEK3 사이트와 효율이 1% 미만인 4개의 잘 editing되지 않는 synHEK3 사이트의 유전체 브라우저 흐름을 보여줌.
[Fig 2G] 이 실험에서 제시한 가설은 Prime Editing이 전사적으로 활발한 영역과 활성 전사 시작점(TSS) 근처에서 가장 효율적임을 의미함. 실험 결과, Prime Editing의 효율성이 유전자의 발현 수준과 TSS에 가까운 위치와 상관관계가 있음을 확인함.
[Fig 2H] Guide와 target 서열의 변동에도 불구하고, editing 효율성은 synHEK3 유래 베타 회귀 모델에 의해 잘 예측됨
[Fig 2I] DeepPrime 점수를 우리 모델과 결합해도 예측 정확도가 더 이상 증가하지 않음
*TSS:transcriptional start sites
Compared editing outcomes for prime vs. Cas9 editing across an identical set of genomic locations.
Prime editing과 Cas9 editing의 편집 결과를 동일한 유전체 위치 세트에서 비교
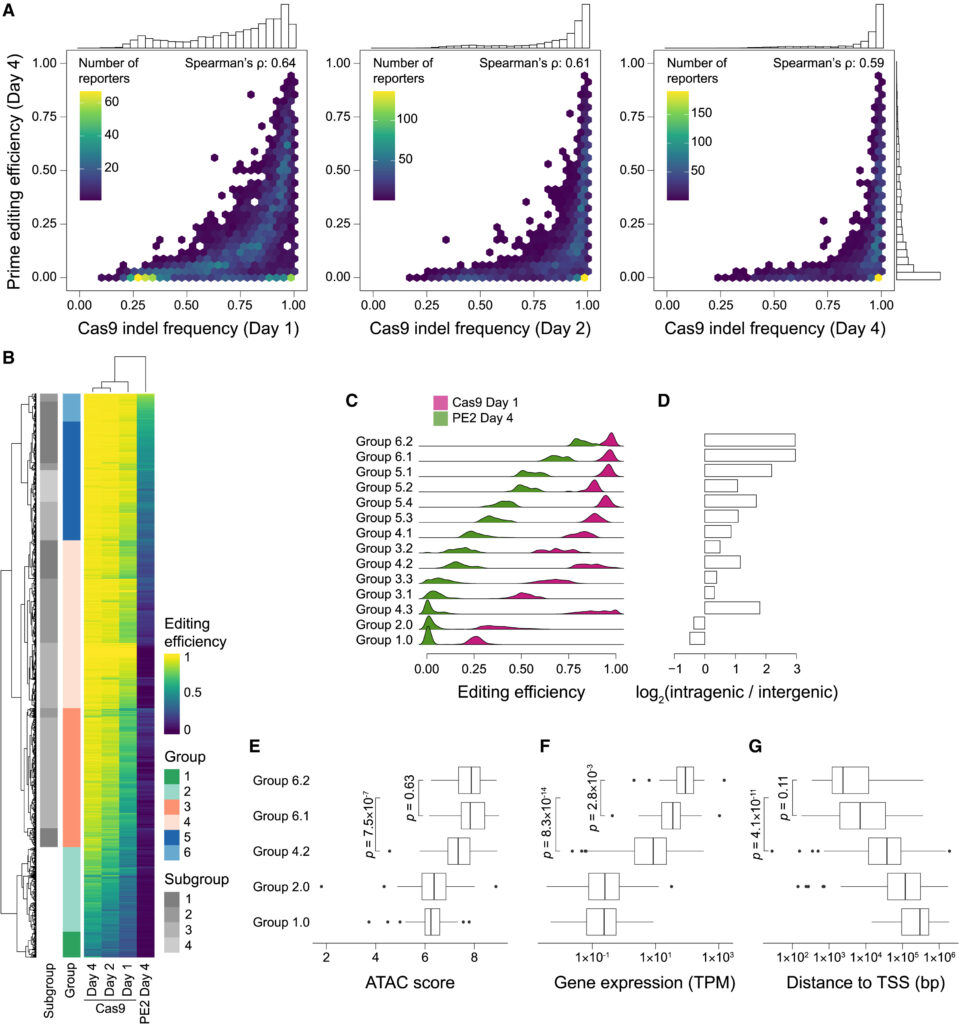
Fig. 3. 우리는 K562 세포주 에서 동일한 HEK3 스페이서 시퀀스를 타겟으로 하는 gRNA를 사용하여 통합된 reporter들의 공통 세트를 활용하여 Cas9와 prime editing 결과를 비교했습니다.
(A) 동일한 synHEK3 리포터 세트에서 Cas9 (전송 후 1, 2, 또는 4일) 대 prime editing(전송 후 4일)의 편집 효율성 비교. 각 bin에 할당된 리포터 수에 따라 색상이 지정된 플롯입니다. 편집 효율성의 1차원 히스토그램이 위쪽과 오른쪽에 표시됩니다.
(B) prime 및 Cas9 editing 효율성에 기반한 synHEK3 리포터들의 계층적 클러스터링.
(C) 14개 그룹의 synHEK3 리포터들에 대한 prime 및 Cas9 editing 효율성 밀도 플롯. 평균 prime editing 효율성에 따라 정렬됩니다.
(D) 각 14개 그룹에서 유전자 내 vs. 유전자 간 사이트 수의 로그2 비율을 보여주는 막대 그래프.
(E–G) 선택된 그룹의 유전자 내 사이트의 특성 비교. p 값: 양측 Kolmogorov-Smirnov 검정. (E) 선택된 그룹의 전사 포즈 창고에 대한 접근 가능한 크로마틴과 시퀀싱 (ATAC-seq) 점수의 상자 그림. (F) 선택된 그룹의 겹치는 유전자의 발현 수준 (TPM; x 축, log10 스케일). (G) 선택된 그룹의 synHEK3 리포터와 가장 가까운 TSS 사이의 거리 (bp; x 축, log10 스케일).
[Fig. 3A] Cas9 편집이 프라임 편집보다 더 효율적이었으며 삽입 및 결실(indels)이 첫날부터 관찰됨. 4일째까지 리포터의 75%에서 삽입 및 결실 빈도가 90%를 초과했으며, Cas9는 prime editing에 저항성을 가진 synHEK3 리포터를 성공적으로 편집
[Fig. 3B] 두 편집 방법의 차이를 탐구하기 위해, 그들은 prime editing과 Cas9 editing의 측정 된 효율성을 기반으로 synHEK3 리포터들을 6개의 그룹으로 클러스터링하여 Heat map을 그림.
[Fig. 3C] 전체적으로 14개의 클러스터를 볼 수 있음. Prime editing과 Cas9 editing 효율성은 일반적으로 상관 관계가 있었지만, 몇몇 그룹은 이 상관 관계를 넘어서는 특성을 보임. 예를 들어, prime editing과 Cas9 모두에서 높은 편집 효율을 보인 그룹(그룹 6.1 및 6.2), Cas9에서는 높지만 prime editing에서는 낮은 효율을 보인 그룹(그룹 4.2 및 4.3), 그리고 두 편집 방식 모두에서 낮은 효율을 보인 그룹(그룹 1.0 및 2.0)을 볼 수 있음.
[Fig. 3D] 그룹 6.1과 6.2는 비슷하게 높은 Cas9 편집 효율을 보였지만(평균: 96.1% 대 96.8%, p = 0.1), 그룹 6.2는 prime editing에서 유의미하게 높은 효율을 보였음(평균: 69.3% 대 82.5%, p = 6.6 × 10⁻¹⁶). 그룹 6.1과 6.2 모두 대부분이 유전자 내부에서 일어나는 현상을 나타냄.
[Fig. 3E-G] 그룹 4.2와 4.3은 높은 Cas9 RNP 편집 효율(평균: 86.4% 및 89.9%)을 보였으나, prime editing 효율은 낮았습니다(평균: 17.1% 및 2.2%). 그룹 4.3의 많은 synHEK3 사이트는 prime editing 효율이 거의 0에 가까웠는데, 이는 PE2 구조의 PB 절제 기술 인공물 때문이라고 판단되었습니다. 그러나 그룹 4.2 사이트는 유전자 내부에 위치하고 접근 가능하지만 발현 수준이 낮고(TSS에서 멀리 떨어져 있음), 이는 Cas9 editing은 가능하지만 prime editing 은 어려운 원인으로 작용합니다.
Investigating the cis-chromatin context dependencies of trans-acting factors influencing prime editing
Prime editing 에 영향을 미치는 trans- 작용 인자의 cis-chromatin 문맥 의존성 조사
Fig. 4. 프라임 에디팅의 크로마틴 문맥 의존적 조절을 수정된 sci-RNA-seq3 워크플로우를 사용해 해부하기
(A) 이 실험에서 사용된 두 개의 단클론 K562 세포주는 PE2와 역 테트라사이클린 전사활성화제(rtTA)를 안정적으로 발현하며, 함께 50개의 고유한 synHEK3 리포터를 포함하고 있습니다. 세포는 높은 감염 다중도(MOI)로 TRE-shRNA 라이브러리와 트랜스듀싱되었으며, 독시사이클린으로 처리되었습니다. 2일째, 세포는 synHEK3 리포터에 무작위 6-bp 삽입을 도입하기 위해 pegRNA로 트랜스펙션되었습니다. 3-4일 후, 핵을 추출하고 고정하였습니다. TRE는 테트라사이클린 반응 요소를 의미합니다.
(B) 고정된 핵은 T7 중합효소(핑크색 원)로 synHEK3 및 shRNA 구성요소로부터 전사체를 생성하는 IST(Indexed Single-cell Transcriptomics) 처리를 거쳤습니다.
(C) 핵은 인덱스된 역전사(RT)를 위해 96웰로 분배되었습니다. 각 wells에서는 oligo-dT 프라이머와 synHEK3 및 shRNA 특이적 프라이머를 포함한 세 가지 인덱스된 RT 프라이머 혼합물을 사용했습니다.
(D) RT 후, 핵은 모아서 인덱스된 헤어핀 연결을 위해 96well 플레이트에 재분배되었습니다. 그 후, 핵은 모아지고 최종 96웰 플레이트로 나누어졌습니다. 이중 가닥 합성 후, 핵은 용해되고 결과물 용해액은 두 개의 플레이트로 나누어졌습니다. 한 플레이트는 전사체 라이브러리를 생성하기 위해 Tn5 태그멘테이션 및 인덱스된 PCR을 거쳤습니다. 다른 플레이트는 synHEK3 및 shRNA 전사체를 표적하는 인덱스된 농축 PCR에 사용되었습니다.
(E) 각 synHEK3 리포터에 대해 편집 결과를 계산하고 특정 shRNA가 있는 세포와 없는 세포 간의 결과를 비교했습니다.
[Fig. 4A] 교란과 prime editing 결과를 동시에 포착하기 위해, 세포는 수정된 sci-RNA-seq3 work flow를 통해 프로파일링함
[Fig. 4B] 첫 번째 변경은 RT 단계 전에 발생함. 즉, 메탄올로 고정된 핵에서 T7 IST를 수행하여 synHEK3 리포터와 shRNA 구성을 통해 RNA 발현을 유도함.
[Fig. 4C] 두 번쨰 변경은 RT 단계에서는 mRNA(oligo-dT), synHEK3, 및 shRNA 전사체를 대상으로 하는 프라이머 혼합물이 사용되어, 주어진 세포에서 전사체, 편집 결과 및 교란을 동시에 캡처 함
[Fig. 4D] 마지막 단계에선 프로토콜이 끝날 무렵에 핵 용액을 두 부분으로 나눔. 한쪽은 기존의 sci-RNA-seq3 라이브러리 준비를 위해 사용되었고, 다른 한쪽은 synHEK3 및 shRNA 유래 전사체를 타겟으로 하는 풍부화 PCR에 사용됨.
[Fig. 4E] 이 행렬에 대해 정량적 형질좌위 분석 스타일의 방법을 적용함. 각 synHEK3-shRNA 쌍에 대해 shRNA를 받은 세포와 받지 않은 세포의 synHEK3 리포터에서의 편집 빈도를 비교
shRNA-mediated knockdown effects on prime editing outcomes
-shRNA 매개 knock down이 prime editing 결과에 미치는 영향을 감지
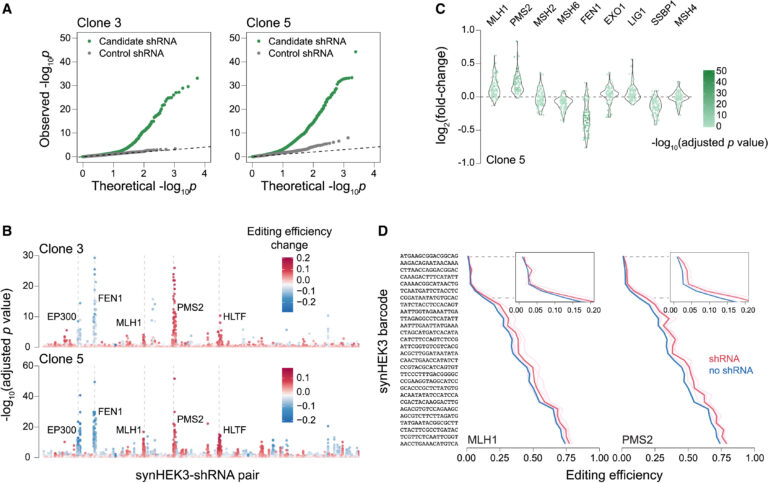
Fig 5. MMR 관련 유전자를 교란했을 때 prime editing 에 미치는 영향
(A) 클론 3(왼쪽)과 클론 5(오른쪽)에서 synHEK3-shRNA 쌍의 통계적 유의성(log10)을 나타내는 분위-분위(Q-Q) 플롯. 후보 shRNA(녹색)와 대조군 shRNA(회색)는 별도로 플롯되었습니다. p 값: 이항 검정.
(B) 모든 synHEK3-shRNA 쌍의 조정된 p 값(log10)을 나타내는 플롯. 높은 통계적 유의성을 가진 표적 유전자가 주석으로 달려 있습니다. 점의 색상은 해당 shRNA에 의해 유도된 편집 효율 변화에 따라 다릅니다.
(C) 클론 5에서 MMR 관련 유전자를 표적으로 하는 shRNA의 효과. synHEK3-shRNA 쌍의 prime editing 효율의 로그2 배 변화를 플롯하고 해당 조정된 p 값(log10)에 따라 색상을 지정했습니다.
(D) MLH1 및 PMS2에 대한 shRNA의 효과. 핑크색 선: 개별 shRNA가 있는 세포에서의 편집 빈도; 빨간색 선: 유전자 표적 shRNA의 평균 편집 빈도; 연파란색 선: 개별 shRNA의 대조군 편집 빈도(평균 선에 비해 분산이 낮아 보이지 않음, 파란색으로 표시); 파란색 선: 대조군 평균 편집 빈도.
[Fig 5A] 두 개의 단클론 라인에서 모든 synHEK3-shRNA 조합을 살펴본 결과, 대조 shRNA를 포함한 쌍에 비해 유의미한 synHEK3-shRNA 쌍이 명확하게 더 많음을 관찰할 수 있었음
[Fig 5B] 여러 유전자를 타겟으로 하는 shRNA들은 두 개의 클론 모두에서 매우 유의미하고 재현 가능한 결과를 보였음.
[Fig 5C] 이 효과를 더 잘 시각화하기 위해, 각 MMR 관련 인자를 타겟으로 하는 synHEK3-shRNA 쌍을 통합하여 보여주었음
[Fig 5D] 가장 강력한 prime editing 촉진 효과는 PMS2와 MLH1을 타겟으로 하는 shRNA에서 관찰되었음
HLTF works as a chromatin context-dependent repressor of prime editing
– HLTF는 염색질 문맥에 의존적으로 프라임 에디팅을 억제하는 역할을 함
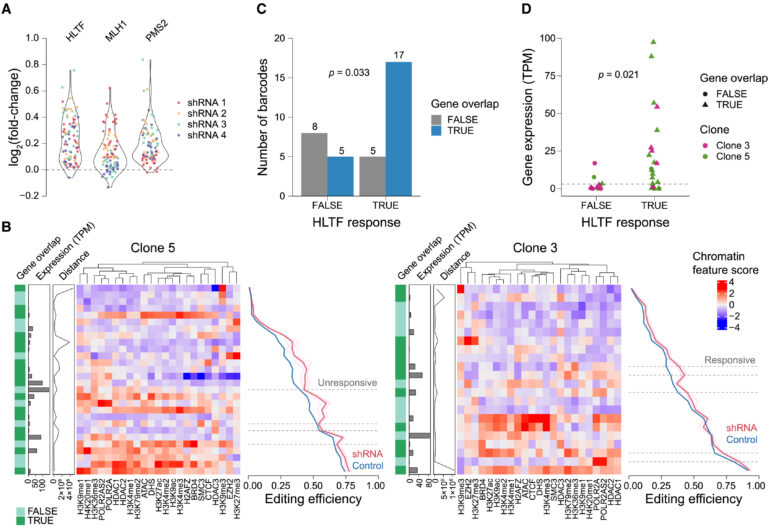
Fig 6. HLTF 억제에 대한 Chromatin context 특이적 반응
(A) HLTF, MLH1, 및 PMS2의 억제에 대한 synHEK3 사이트의 편집 효율성의 변화에 대한 바이올린 플롯. 포인트 점은 shRNA의 정체성에 따라 색상이 다릅니다.
(B) synHEK3 리포터(행)와 HLTF에 대한 shRNA의 반응(왼쪽: 클론 5; 오른쪽: 클론 3)의 히트맵. 왼쪽 끝 바는 synHEK3 리포터와 GRCh38 유전자 주석의 중첩 상태를 주석합니다.
두 번째 왼쪽 바는 중첩된 또는 가장 가까운 유전자의 TPM에서의 발현 상태를 나타냅니다. 세 번째 왼쪽 바는 유전자-중첩 synHEK3 리포터 또는 유전자 밖의 synHEK3 리포터에 대한 TSS까지의 거리(bp)를 나타냅니다. 중앙 히트맵은 synHEK3 리포터의 스케일된 크로마틴 특성 점수를 사용하여 생성되었으며 열에 따라 클러스터링되었습니다.
오른쪽 선 플롯은 HLTF에 대한 shRNA의 효과를 보여줍니다. 분홍색 선: 개별 shRNA를 가진 세포에서의 편집 빈도; 빨간 선: 유전자 표적 shRNA의 평균 편집 빈도; 연한 파란색 선: 개별 shRNA의 대조군 편집 빈도(평균 선에 비해 변동이 낮아 보이지 않음, 파란색으로 표시됨); 파란색 선: 대조군 편집 빈도의 평균. 점선: HLTF 억제에 대한 차별적인 반응을 보이는 사이트.
(C) HLTF 억제에 대한 반응성과 GRCh38 유전자 주석과의 중첩 상태에 따라 synHEK3 리포터 수를 나타낸 막대 플롯. p 값: Fisher의 정확 검정.
(D) synHEK3 리포터와 중첩되거나 근처(10 kb 이내)에 있는 유전자들의 발현. 점선 위의 유전자(TPM = 3)는 발현된 것으로 간주됩니다. p 값: 양측 Kolmogorov-Smirnov 검정.
[Fig 6A] 이전 연구에서는 HLTF를 억제하면 prime editing에 의한 SNP 빈도가 약간 감소한다고 보고됨. 반면, 우리는 HLTF 억제 시 6-bp 삽입이 강하게 증가하는 것을 관찰했으며, 이는 MLH1 및 PMS2를 타겟으로 하는 shRNA로 유도된 변화와 유사한 것을 알 수 있음.
[Fig 6B] 개별 synHEK3 삽입 부위를 검사한 결과, HLTF 억제에 대한 이질적인 반응을 heat map pattern에서 볼 수 있
[Fig 6C] 그들은 선택된 synHEK3 리포터들을 HLTF shRNA Knock down에 대한 반응성에 따라 그룹화 하고, 주석이 달린 유전자와의 중복 상태를 계산함. 반응성 그룹은 유전자 중복 부위에서 풍부하게 나타남.
[Fig 6D] 더 나아가 중복되거나 인접한 유전자(10 kb 이내)의 발현 상태를 고려했을 때, 반응성 부위는 주로 활발히 전사 되는 유전자 근처에 위치한 반면, 비반응성 부위는 대부분 비전사 영역에 위치함.
Modulating prime editing outcomes through targeted epigenetic reprogramming and enhancing prime editing efficiency by transient gene activation
–표적 후성 유전학적 재프로그래밍을 통해 prime editing 결과를 조절하고, 일시적인 유전자 활성화를 통해 prime editing 효율을 증가
Figure 7. 후생유전적 조절을 통한 프라임 편집 결과의 조절
(A) CRISPRoff 실험의 워크플로우.
(B) USP7, METTL2A, 및 LRRC8C 프로모터를 타겟으로 하는 CRISPRoff gRNA가 도입된 세포에서 synHEK3 리포터의 평균 프라임 편집 효율성의 산점도. 해당 타겟 유전자에 있는 synHEK3 리포터는 레이블이 붙어 있습니다. 오차 막대는 측정된 편집 효율성의 표준 편차를 나타냅니다.
(C) CRISPRoff 실험에서 synHEK3 리포터의 프라임 편집 효율성 변화에 대한 막대 플롯. 대조군 편집 효율성은 (B)에서 보여준 CRISPRoff 타겟 유전자에 포함되지 않은 synHEK3 리포터를 이용해 훈련된 선형 모델을 사용하여 예측된 효율성입니다.
(D) CRISPRa 실험의 워크플로우.
(E) K562 세포에서의 내인성 유전자 타겟에 대한 프라임 편집 효율성(%), CRISPRa를 통한 후생유전적 편집 여부에 따른 결과.
녹색 막대는 PEmax와 (e) pegRNA만을 도입한 와일드타입 K562 세포 라인에서의 평균 프라임 편집 효율성을 나타냅니다. 회색 막대는 대조군 프로모터가 CRISPRa를 통해 활성화된 경우의 평균 프라임 편집 효율성입니다. 파란색 막대는 타겟 유전자 프로모터가 활성화된 경우의 평균 프라임 편집 효율성입니다. CRISPRa (파란색)와 대조군 (회색) 그룹 간의 비율 변화가 계산되었습니다. 인셋에는 첫 세 유전자에 대한 확대 뷰가 표시되어 있습니다.
(F) 인간 iPSC 라인에서의 내인성 유전자 타겟에 대한 프라임 편집 효율성(%), CRISPRa를 통한 후생유전적 편집 여부에 따른 결과. 회색 막대는 대조군 프로모터가 CRISPRa를 통해 활성화된 경우의 평균 프라임 편집 효율성입니다. 파란색 막대는 타겟 유전자 프로모터가 활성화된 경우의 평균 프라임 편집 효율성입니다.
(G) IL2RB 외온의 컨트롤 세포와 CRISPRa 세포에서 측정된 평균 prime 에디팅 효율의 산점도입니다. 점은 편집 유형에 따라 색상으로 구분되어 있으며, x축과 y축은 로그10 스케일입니다. 분홍색과 빨간색 선은 CRISPRa 그룹과 컨트롤 그룹 간의 2배, 5배, 10배 차이를 나타냅니다.
(H) 모든 변형에 대한 프라임 에디팅 효율의 fold-change를 나타낸 상자 그림입니다. 점선은 2배의 fold-change를 나타냅니다.
[Fig 7A] 이 세포들을 epidemic editing 도구인 CRISPRoff-v2 및 이들 유전자의 promotor 를 침묵 시키기 위한 gRNA 쌍으로 일시적으로 형질 도입한 후, synHEK3 리포터 전반에 걸쳐 prime editing효율을 측정함.
[Fig 7B] 표적 유전자 외부의 synHEK3 리포터에 대해 보정함
[Fig 7C] USP7, METTL2A, 및 LRRC8C 유전자에 있는 synHEK3 리포터의 에디팅 빈도가 각각 39%, 26%, 및 47% 감소한 것을 관찰할 수 있었음.
[Fig 7D] dCas9-VP64 융합 단백질을 안정적으로 발현하는 K562 세포주에 표적 유전자의 promotor를 Targeting하는 gRNA 쌍(2XMS2)과 MCP-p65-Rta 융합 단백질을 형질 도입함 그 후 2일 후 원하는 돌연변이를 프로그래밍하기 위해 PEmax와 pegRNA를 세포에 형질 도입함.
[Fig 7E] 동일한 (e)pegRNA의 에디팅 효율을 WT K562 세포주에서 측정한 결과, 유사한 기준선을 관찰하였으며, 이는 프라임 에디팅이 CRISPRa 기구에 의해 억제되지 않는다는 것을 보여줌
[Fig 7F] 세포 환경에서 효과적인지 테스트하기 위해, 인간 iPSC에서 CXCR4, WNT3A, 및 EGFR의 prime editing을 CRISPRa로 선행한 후 각각 7.8배, 1.7배, 및 1.5배의 editing 효율 증가를 관찰.
[Fig 7G] Small insertion(1, 3, 6 bp)은 CRISPRa에 가장 민감하게 반응함.
[Fig 7H] 단일 뉴클레오타이드 치환 중에서는 G/A 및 A/G 편집이 CRISPRa에 가장 민감하게 반응한 것을 알 수 있다
Disscussion
이 연구는 T7 프로모터를 포함한 간단한 리포터 구성체와 T7 IVT 및 IST를 활용하여 염색질 위치 효과를 대량 또는 단일 세포 형식으로 분석하는 방법을 제시합니다. 대량 분석에서는 T7 IVT를 통해 밀집된 리포터의 거의 완전한 매핑과 위치 의존적 조절 효과 측정이 가능하였고, 단일 세포 RNA-seq에서는 T7 IST로 비전사 유전체 구성체를 동시에 프로파일링했습니다. 이 방법은 19-bp T7 프로모터를 리포터에 추가하는 것만으로 적용 가능하며, 무작위로 통합된 리포터 또는 작용제 구성체의 정확한 유전체 좌표를 포착하는 데 유용합니다. 연구팀은 4,000개 이상의 유전체 위치에서 프라임 에디팅 효율을 측정하고, 23개의 염색질 특징과의 상관관계를 분석했습니다. 베타 회귀 모델링을 통해 H3K79me2가 프라임 에디팅의 주요 예측 변수임을 밝혔으나, 이는 활성 전사와의 강한 상관관계 때문일 가능성이 높습니다. 이 모델은 또한 내인성 유전체 표적 사이트를 타겟팅하는 epegRNA의 편집 효율을 예측할 수 있는 유용한 도구로 나타났습니다. 전사 작용 인자와 염색질 환경의 상호작용을 조사한 결과, HLTF가 문맥 의존적인 prime editing억제제로 작용하며, HLTF를 노크다운하면 활발히 전사되는 부위에서 프라임 에디팅이 선호적으로 향상되었습니다. HLTF의 기능이 직접적인 영향을 미친다기보다는 DNA 수리에서의 역할과 관련이 있을 가능성이 높습니다. 이를 통해 HLTF의 역할과 관련된 추가 연구가 필요함을 제시합니다. CRISPRa 조건화가 유전자 내부 프라임 에디팅의 효율을 향상시킬 수 있음을 보여주었습니다. CRISPRoff를 이용한 유전자 침묵이 프라임 에디팅 효율을 감소시키는 반면, CRISPRa로 먼저 “조건화“하면 내인성 표적의 편집 효율이 현저히 향상됩니다. 이 방법은 K562 및 iPSC 세포주에서 유효성을 검증했으며, CRISPRa 조건화가 다양한 편집 유형에서 강력한 향상을 가져온다고 보고합니다.
몇 가지 이 연구의 한계를 논의 하자면 첫째, 23개의 염색질 특징만으로는 epigenome의 모든 측면을 포착하기 어려울 수 있으며, distal intergenic 영역의 synHEK3 리포터들이 높은 변동성을 보이는 것은 ENCODE 분석이나 고차 구조 이상의 이질성 때문일 수 있습니다. 둘째, 초기 실험에서 단일 pegRNA를 사용해 같은 삽입을 고정된 표적 사이트에 도입하였으나, 돌연변이 유형, 표적 서열, cis-염색질 환경, 세포 유형 간의 상호작용을 고려하지 않았습니다. 셋째, shRNA 단일 세포 스크리닝에서 prime editing 효율에 대한 교란의 효과 크기가 Repair-seq에서 측정 된 것보다 작았으나, 이는 shRNA의 비효율적인 knock down 때문이 아니라 6-bp 삽입의 생리학적 조절 때문일 가능성이 높습니다. 마지막으로, 후성 유전학적 리프로그래밍을 적용할 때, 편집기 간의 간섭 가능성이 우려됩니다. CRISPRa 기구를 지속적으로 발현하는 세포에서는 일관된 prime editing 억제 현상이 관찰되지 않았으나, CRISPRa와 prime editing 표적 사이트가 너무 가까운 경우에는 간섭이 발생할 수 있습니다. 다른 epigenome 공학 도구나 일시적으로 존재하는 CRISPR RNP를 사용하면 서로 다른 CRISPR 매개 모듈 간의 교차 간섭을 줄일 수 있습니다.