희귀한 copy-number variation이 인간의 복잡한 형질에 미치는 영향
인간 게놈은 copy-number variants (CNV)를 품고 있는 수십만 개의 영역을 포함하고 있습니다. 그러나 큰 biobanks에 의해 생성된 SNP-array 데이터에서 더 큰 CNV만 확인 가능했기 때문에 대부분의 polymorphism의 phenotype effect는 알려져 있지 않습니다. 이 연구에서는 CNV를 더 민감하게 감지하기 위해 biobank cohort의 haplotype 공유를 활용하는 계산 접근법을 개발했습니다. UK Biobank에 적용된 이 접근법은 게놈 구조 변화에 의해 생성된 모든 희귀 유전자 불활성화 사건의 약 절반을 차지했습니다. 이 CNV call set를 통해 CNV와 56개의 정량적 특성 사이의 연관성에 대한 자세한 분석이 가능했으며, CNV에 의해 인과적으로 구동될 가능성이 있는 269개의 독립 연관성(p < 5 × 10-8)을 식별했습니다. 추정된 표적 유전자는 절반 정도의 loci에서 식별 가능하여, 이러한 유전자의 용량 민감도에 대한 통찰을 가능하게 하고 몇 가지 유전자 특성 관계를 밝혀냈습니다. 이러한 결과는 인간이 지니는 복잡한 형질의 유전적 기초에 대한 통찰력을 제공하는 haplotype 정보 분석의 능력을 보여줍니다.
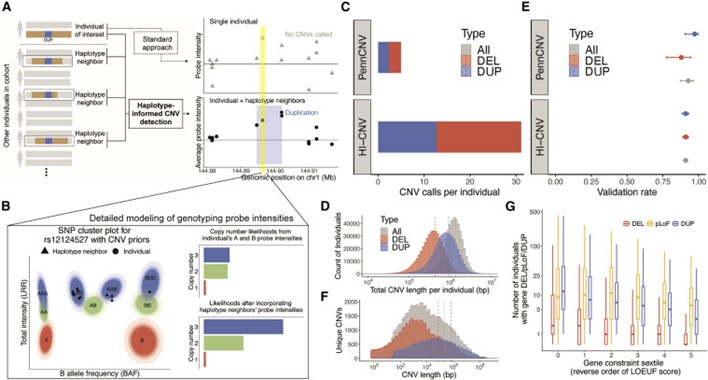
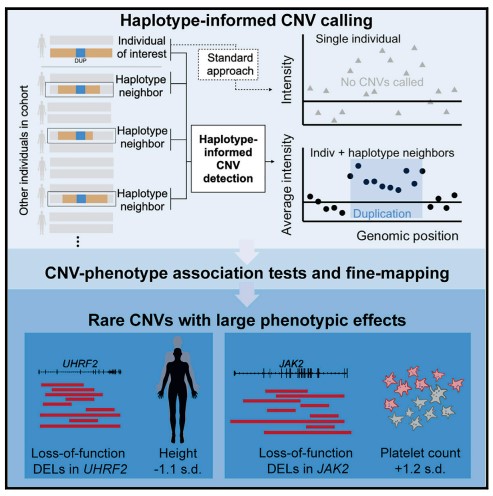
[Figure 1] UK Biobank의 SNP-array data 에서 haplotype에 대한 정보를 제공하는 CNV 탐지
(a) HI-CNV framework는 오랫동안 공유된 haplotypes (인접한 haplotype)이 있는 개인의 해당 데이터와 함께, 개인의 SNP-array data를 분석하여 CNV를 탐지하는 능력을 제공합니다. 이와는 대조적으로, 표준 접근법은 개인의 데이터를 단독으로 분석합니다.
(b) SNP-specific genotype cluster는 map allele-specific(A 및 B allele) probe 강도 측정을 copy-number likelihood에 대한 확률적 정보에 비해 prior로 둡니다.
(c) PennCNV 및 HI-CNV를 통해 계산된 CNV의 평균량. 각 UK Biobank 참가자에 대해 계산되었습니다.
(d) 개인 당 전체 CNV 길이의 분포 (HI-CNV call set)
(e) PennCNV 및 HI-CNV에서부터 측정된 CNV call의 validation rate. 43명의 UK Biobank 참가자에 대해 측정되었으며, 각각 독립적인 whole-genome 시퀀싱 데이터를 보유합니다. 에러바는 95% CI를 나타냅니다.
(f) CNV 길이들의 분포 (HI-CNV call set)
(g) n = 452,500 UK Biobank 참가자들에 대한 whole-gene deletion 및 duplication 및 pLoF CNV의 분포(gene set은 두드러지게 통제된 것들을 사용). Box plot의 중앙은 중간값을 의미하며, 박스의 위아래는 각각 75%, 25%를 의미합니다. 그 밑과 위로는 5%, 95%까지 표현했습니다.
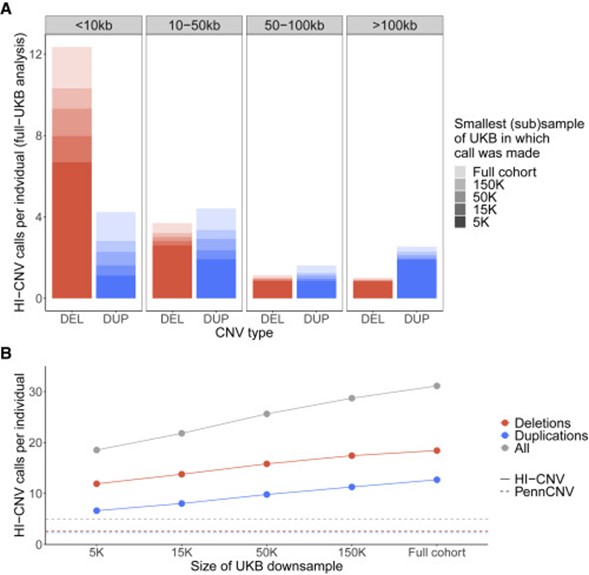
[Figure 2] UK Biobank 데이터셋의 하위 샘플들에 대한 HI-CNV 성능 벤치마크 결과
더 작은 샘플 크기에서도 HI-CNV가 감지 감도를 향상시키는 정도를 평가하기 위해, 연구진들은 UK Biobank의 다양한 하위 샘플에서 HI-CNV의 성능을 벤치마킹했습니다. (N = 5K, 15K, 50K, 150K)
(a) 모든 하위 샘플에 포함된 500명의 개인에 대해, 전체 N ~ 500K에 대한 분석에서 해당 개인들에서 수행된 각 CNV call에 대해, 이들은 call이 감지된 최소 샘플 (n = 5K, 15K, 50K, 150K, or full cohort)을 결정했습니다. 전체 막대 높이는 위에 명시한 이벤트의 크기 및 CNV 유형(삭제 대 중복)으로 분류된 500명의 개인(전체 N ~ 500K 분석에서)에 걸친 평균 CNV call 수를 나타냅니다. 음영은 각 호출이 처음 감지된 하위 샘플(하위 샘플에서 지정된 호출을 겹치거나 완벽하게 복제하는 호출로 정의됨)을 반영합니다. 이러한 분석은 검출 민감도가 예상대로 샘플 크기에 따라 증가했지만(특히 10kb 미만의 작은 CNV에 대해) 전체 UK Biobank cohort를 사용하여 이루어진 대부분의 CNV 호출은 n = 5K의 샘플 크기에서 HI-CNV에 의해 이미 검출되었음을 보여주었습니다.
(b) HI-CNV (N = 5K, 15K, 50K, 150K, 전체 샘플)와 PennCNV의 개인당 평균 call 수를 비교했습니다. 개인 당 CNV call의 평균 수는 CNV 유형에 따라 다양한 하위 표본에 걸쳐 표시됩니다. 수평선은 전체 UK Biobank cohort에 걸쳐 PennCNV가 감지한 평균 이벤트 수를 반영합니다. (각 하위 샘플에서 call [89%–93%]은 전체 코호트를 사용하여 한 call을 복제하거나 중복하여, 위처럼 샘플 수를 줄인 분석에서 효과적인 false-positive 제어를 나타냅니다.)
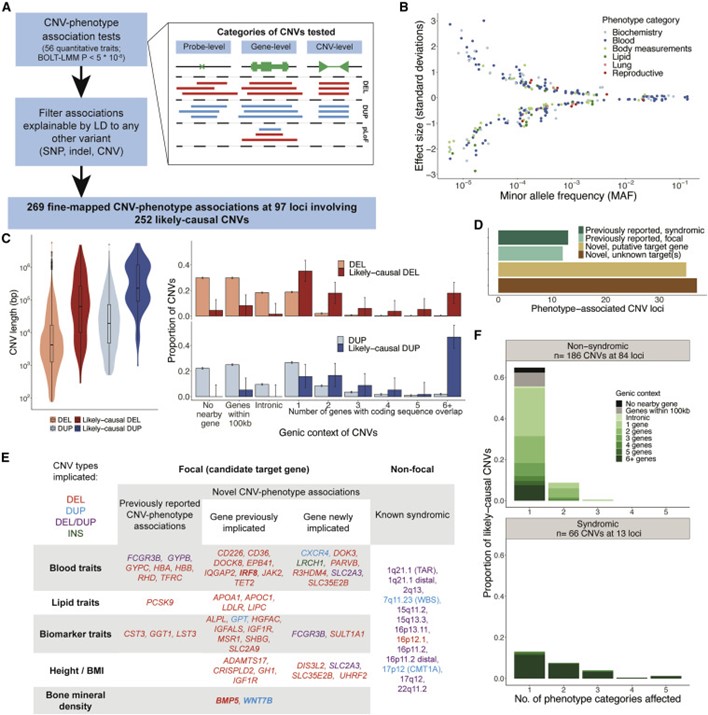
[Figure 3] Fine-mapping analysis를 통해 likely causal CNV-trait 연관성 파악
(a) 연관성 및 fine-mapping 연구의 파이프라인. 옆에 포함된 그림은 테스트한 CNV의 세 종류를 이야기합니다.
(b) likely-causal한 269개의 CNV-phenotype 연관성에 대한 Effect size와 minor allele frequency를 그린 plot. 각 phenotype 카테고리에 따라 색을 다르게 표현했습니다.
(c) CNV 길이의 분포(왼쪽), 유전적 특징(오른쪽). 모든 CNV에 대해, 그리고 likely causal CNV에 대해 각각 표현했습니다.
(d) 기존 연구들에서 제시된 기준 및 추정된 target gene에 대한 97개의 CNV loci를 보다 쪼개어 확인한 결과입니다.
(e) 후보 표적 유전자는 (1) CNV-phenotype 연관성이 이전에 보고되었는지, (2) 표적 유전자는 기존 보고된 코딩 변이 연관성 또는 이전 실험 연구에서 지목되었는지, 또는 (3) 위의 둘 중 어느 쪽도 아닌지 여부에 따라 분류되었습니다. 맨 오른쪽 열에는 여기서 다시 식별된 syndromic CNV가 나열되어 있습니다. 색은 CNV 유형을 나타내고 굵은 글꼴은 잠재적으로 표적 유전자를 조절하는 non-coding CNV를 나타냅니다.
(f) CNV와 관련된 phenotype 카테고리의 수에 따라 분류된 syndromic CNV(하단)와 non-syndromic CNV(상단)의 유전적 맥락입니다.
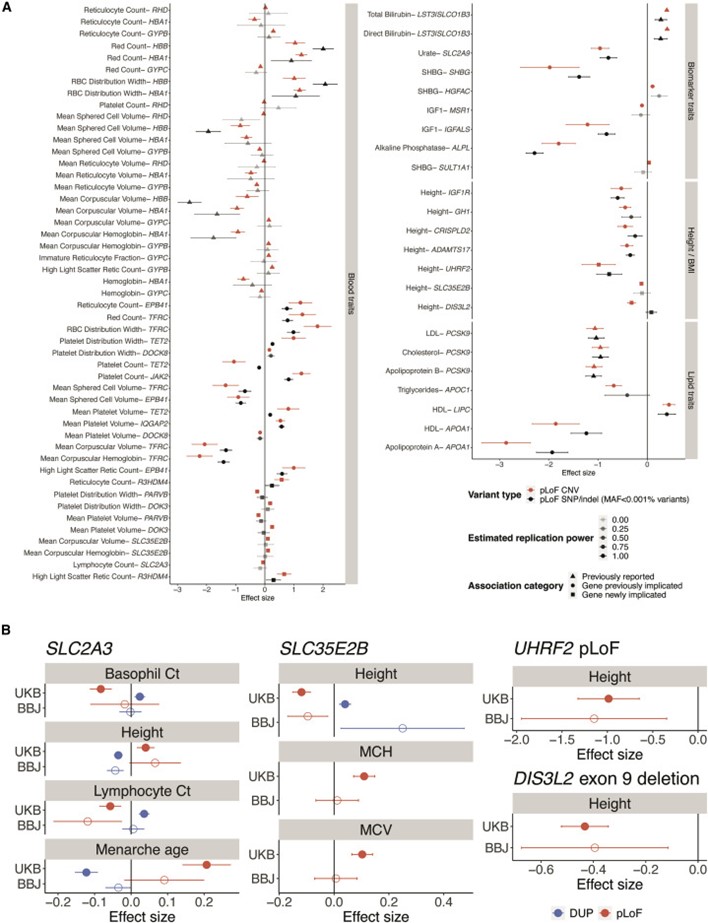
[Figure 4] CNV-phenotype 연관성의 확인 및 재현
(a) UK Biobanks의 loss-of-function burden 분석. 중점적인 후보 표적 유전자(figure 3E)에 작용한다고 알고 있던 CNV와 관련된 연관성에 대해, 추정 표적 유전자의 loss-of-function(pLoF)을 일으킬 것으로 예측된 CNV의 추정된 효과를 동일한 유전자의 초희소 pLoF SNP 및 indel 변이의 추정된 효과와 비교했습니다(최근 UK Biobank whole-exome 분석에서 보고됨: SNP/indel pLoF 부담 실험을 수행; Backman et al., 2021). 효과 크기와 95% 신뢰 구간은 pLoF CNV의 경우 빨간색으로 표시했고 pLoF SNP/인델 부담의 경우 검은색으로 표시했습니다. pLoF SNP/indel 부담에 대한 마커와 오류 막대는 연관성을 감지하는 유의성에 따라 음영 처리됩니다(PloF CNV와 동일한 효과 크기를 가정했으며, PloF SNP 및 indel의 통합적인 allele frequency에 대한 설명). 이전에 보고된 연관성은 삼각형으로 표시되고, 이전에 관련된 유전자는 원으로 표시되고, 나머지 유전자는 사각형으로 표시됩니다.
(b) BioBank Japan에서 CNV-phenotype 연관성을 다시 실험해 본 결과입니다. UK Biobank 분석에 의해 추정적으로 밝혀진 유전자 특성 관계에 관련된 14개의 연관성(Biobank Japan에서 사용 가능한 표현형과 유의성을 기반으로 선택)을 재현하려고 시도했습니다. 효과 크기 및 95% 신뢰 구간은 pLoF CNV의 경우 빨간색으로 표시되고 전체 유전자 중복의 경우 파란색으로 표시됩니다.
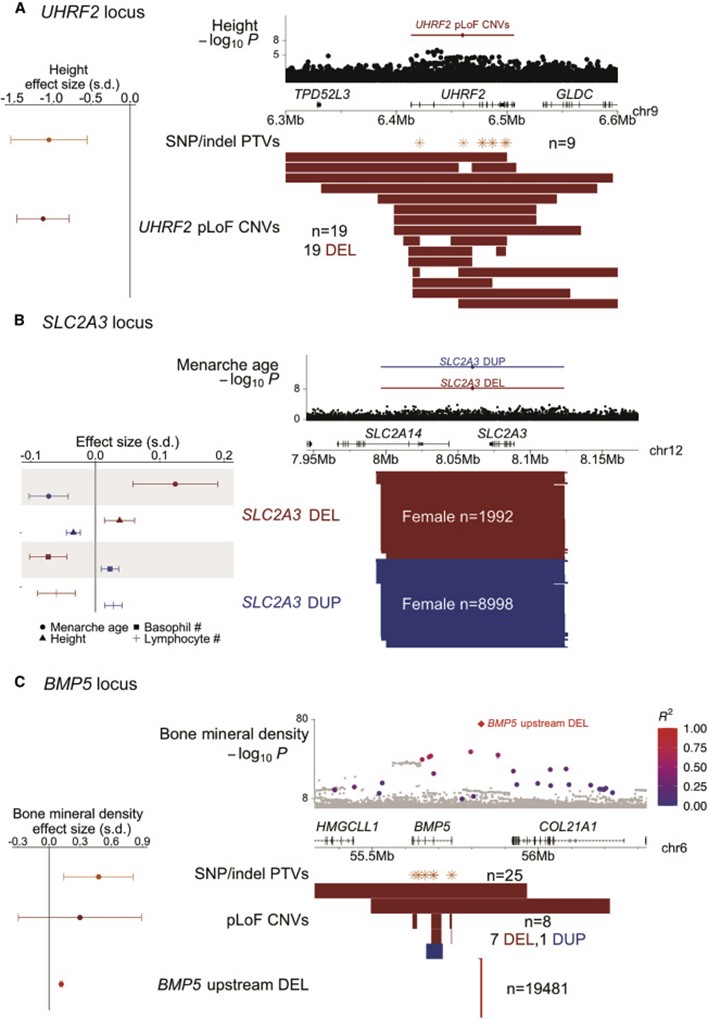
[Figure 5] CNV-phenotype 연관성은 인근 SNP의 연관성보다 강함
(a) UHRF2 locus. (위) UHRF2 pLoF CNV와 근처 SNP들이 키와 가지는 연관성. (아래) UHRF2 pLoF CNV 및 SNP 및 indel PTV의 위치; (왼쪽) 키에 대한 effect size
(b) SLC2A3 locus. (위) SLC2A3 duplication, deletion 및 근처 SNP들이 초경 나이와 가지는 연관성. (아래) SLC2A3 deletion 및 duplication의 위치; (왼쪽) 초경 나이, 키, 호염기성 세포 및 림프구에 대한 effect size
(c) BMP5 locus. (위) BMP5의 upstream deletion이 뼈 미네랄 밀도와 가지는 연관성(R2 > 0.1 인deletion, SNP과 함께 linkage disequilibrium 분류에 따라 색칠되었습니다.) (아래) upstream deletion, BMP5 pLoF CNV, SNP 그리고 indel PTV의 위치. (왼쪽): 뼈 미네랄 밀도에 대한 효과 크기. 모든 패널에서 deletion은 빨간색으로 표시되고 duplication은 파란색으로 표시됩니다. 효과 크기에 대한 오류 막대 및, 95% CI을 표현했습니다. 수치로 나타낸 결과는 Table S5에서, signal intensity plot의 예시는 Figure S3에 상세히 있습니다.
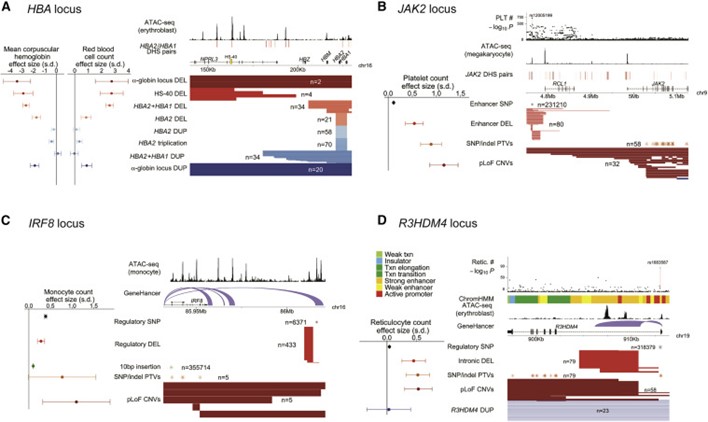
[Figure 6] regulatory 및 gene-altering CNV에 대한 allelic series
(a) HBA locis. α-globin locus에서 CNV의 8가지 종류와 평균 혈구 헤모글로빈 및 적혈구 수에 대한 효과 크기를 나타냅니다. 게놈 주석은 적혈 모세포(Ulirsch et al., 2019), HBA2/HBA1에 대한 distal DNAase I 과민성 부위(DHSs) (Thurman et al., 2012)에서 접근 가능한 크로마틴 영역을 나타내며, HS-40 super-enhancer를 강조합니다.
(b) JAK2 locus. 네 가지 유형의 변이(JAK2 pLoF CNVs, JAK2 SNP 및 indel PTVs, distal enhancer deletion, enhancer 내 공통 SNP rs12005199) 및 혈소판 수에 대한 효과 크기를 나타냅니다. 게놈 주석은 거대핵구(Ulirsch et al., 2019)와 JAK2 distal DHS 쌍(Thurman et al., 2012)에서 접근 가능한 크로마틴 영역을 나타내며, 이는 JAK2의 상류의 220kb에서 공통-SNP 혈소판 수 연관성(상단)으로 국소화됩니다.
(c) IRF8 locus. IRF8 위치에서 미세하게 매핑된 공통 변종과 희귀한 pLoF 변종 및 단핵구 수에 대한 효과 크기를 나타냅니다(putatively regulatory distal deletion, IRF8 pLoFCNV, IRF8 SNP 및 indel PTV). 게놈 주석은 downstream regulatory region 및 IRF8 사이의 단핵구(Ulirsch et al., 2019)와 GeneHancer 연결(Fishilevich et al., 2017)에서 접근 가능한 크로마틴 영역을 나타냅니다.
(d) R3HDM4 locus. 희귀한 CNV, SNP 및 indel PTV, 그리고 R3HDM4의 일반적인 intronic SNP 및 reticulocyte 수에 대한 효과 크기를 나타냅니다. 게놈 주석은 ChromHMM(Ernst, Kellis, 2017) 주석, 적혈 모세포의 접근 가능한 크로마틴 영역(Ulirsch et al., 2019) 및 GeneHancer 연결(Fishilevich et al., 2017)을 나타내며, 모두 R3HDM4의 첫 번째 인트론에서의 조절 기능을 나타냅니다. Lead 관련 SNP rs1683587(상단)도 이 인트론 내에 있어 규제 기능을 시사합니다. (a)와 (b)에서 DHS 쌍은 각각의 상관관계에 따라 색칠되었으며, 옅은 빨간색(연관성 < 0.8)에서 어두운 빨간색(연관성 > 0.95)까지 표현되었습니다. 효과 크기에 대한 오류 막대 및, 95% CI을 표현했습니다. 수치로 나타낸 결과는 Table S5에서, signal intensity plot의 예시는 Figure S3에 상세히 있습니다.
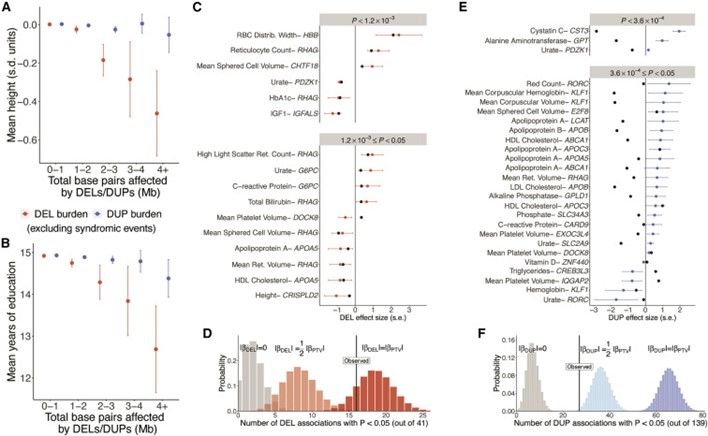
[Figure 7] deletion 및 duplication의 phenotype 효과의 비교
(a, b) 평균 키(a) 및 교육 정도(b)를 deletion 및 duplication에 영향받은 총 게놈 길이와의 관계로 나타냈습니다. 기존에 알려진 syndromic CNV를 포함한 인원은 분석에서 제외되었습니다.
(c) 이전에 trait-altering의 가능성이 있는 PTV(Barton et al., 2021)와 HI-CNV call set가 적어도 두 개의 whole-gene deletion를 포함하는 41개의 유전자 특성 쌍에 대한 표적 분석에서 whole-gene deletion와 정량적 특성 사이의 연관성을 나타냅니다. Whole-gene deletion이 명목상 유의한 연관성을 보인 16개 유전자에 대해 효과 크기와 95% 신뢰 구간이 빨간색으로 표시되었고(p < 0.05), SNP 또는 indel PTV에 대한 효과 크기는 검은색으로 표시되었습니다(Barton et al., 2021).
(d) 16개의 명목상 유의한 연관성을 관찰한 결과, PTV와 동일한 영향을 미치는 whole-gene deletion과 일치했습니다. 확률 분포는 전체 유전자 삭제가 아무런 영향을 미치지 않는 시뮬레이션에서 중요한 연관성 수(회색), PTV의 절반 효과 크기(연분홍색) 또는 PTV와 동일한 효과 크기(빨간색)를 가진다는 것을 나타냅니다.
(e, f) 139개의 gene-trait 쌍을 대상으로 한 표적 분석에서 whole-gene duplication에 대한 유의한 결과 27개가 도출되었으며(p < 0.05), PTV에 비해 효과 크기가 절반 이하인 whole-gene duplication과 일치합니다. (DOCK8 PTV rs192864327에 대한 DOCK8 deletion 및 duplication의 이상 효과 방향은 이 변이에 의해 설명되며 여러 script 중 하나에서만 loss of function을 유발합니다.)
Discussion
위 결과들은 확장된 SNP haplotype를 공유하는 개인에 의해 공유되는 변형에 대한 정보를 모으기 위해 대규모 biobank 코호트 내에서 만연한 원격 관련성을 활용하는 haplotype-informed structural variant 분석의 힘을 보여줍니다. UK Biobank에서 CNV-phenotype 연관성을 탐구하기 위해 적용된 이 접근법은 유전적 변이가 복잡한 특성에 영향을 미치는 다양한 방법들을 밝혔습니다. 몇몇 loci에서, 큰 효과를 가지는 CNV는 추정 표적 유전자를 발견했고, 몇몇 다른 loci에서는 근처의 SNP와 함께 CNV가 SNP analog 없이 기능적 효과를 생성하는 능력을 보여주는 긴 대립 유전자 시리즈를 만들었습니다(예: gene copy-gain 및 regulatory element deletion 혹은 duplication).
여기에 보고된 특정 생물학적 발견 외에도, 우리의 연구는 대규모 게놈 영역에 걸쳐 있을 수 있는 호출하기 어려운 구조 변형에 대한 연관성 및 미세 매핑 분석의 통계적 미묘함을 다루기 위한 신중한 분석 접근 방식을 제공합다. 또한 국소적으로 납 연관성을 나타내는 여러 CNV의 관찰은 SNP 연관성의 통계적 미세 매핑을 수행할 때에도 구조적 변동을 고려하는 것의 중요성을 강조합니다(Beyter et al., 2021; Mukamel et al., 2021).
또한 이러한 결과는 이번 분석에서 접근할 수 없었던 훨씬 더 큰 CNV 세트에 대한 추가 탐색에 동기를 부여할 것입니다. 이들의 접근 방식은 UK Biobank에 대한 이전 분석보다 6배 더 많은 CNV의 탐지를 가능하게 했고, 이러한 CNV는 구조 변동에서 발생하는 것으로 추정되는 희귀 LoF의 약 절반을 차지하는 것으로 보였지만(Collins et al., 2020), SNP-array 데이터에서 검출한 CNV는 각 인간의 게놈이 흔히 존재하는 수천 개의 CNV 중 극히 일부에 불과합니다. (Abel et al., 2020; Collins et al., 2020). 이후 short- & long-read 시퀀싱 데이터를 분석하는 향후 연구가 CNV의 표현형 결과에 대한 더 많은 통찰력을 제공할 것으로 기대합니다.