일반적인 single-nucleotide polymorphism(SNP)은 사람 키의 표현형 변이의 40-50%를 총체적으로 설명할 것으로 예측되지만, 특정 variants와 관련 영역을 식별하려면 큰 표본 크기가 필요하다.
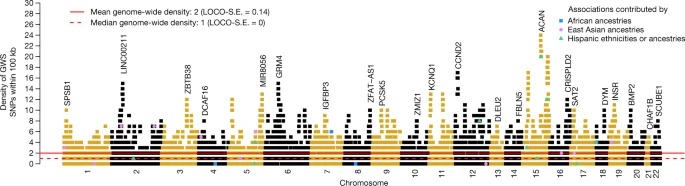
여기서, 우리는 다양한 조상의 540만 개인에 대한 게놈 전체 연관 연구의 데이터를 사용하여 키와 크게 연관된 12,111개의 독립 SNP가 일반적인 SNP-based heritability의 거의 모든 것을 차지한다는 것을 보여준다. 이러한 SNP는 평균 크기가 약 90kb인 중복되지 않는 7,209개의 게놈 세그먼트에 클러스터링되어 있으며, 게놈의 약 21%를 커버한다. 독립적인 결합의 밀도는 게놈에 따라 다양하며, 밀도가 증가하는 영역은 생물학적으로 관련된 유전자에 대해 풍부해진다. 표본 외 추정 및 예측에서 12,111 SNP(또는 HapMap 3 패널)는 유럽 조상의 인구에서 표현형 분산의 40%(45%)를 차지하지만 다른 조상의 인구에서는 약 10-20%(14-24%)만 차지한다. Effect size, 관련 영역 및 유전자 우선 순위는 조상 전반에 걸쳐 유사하며, 이는 감소된 예측 정확도가 관련 영역 내의 연결 불균형 및 대립 유전자 빈도 차이로 설명될 가능성이 있음을 나타낸다. 마지막으로, 우리는 causal gene와 변형을 연루시키는 데 필요한 것보다 더 작은 샘플 크기로 관련 생물학적 경로가 탐지 가능하다는 것을 보여준다.
전반적으로, 이 연구는 일반적인 키 관련 변형의 대부분을 포함하는 특정 게놈 영역에 대한 포괄적인 지도를 제공한다. 비록 이 지도가 유럽 조상의 인구에 대해 포화 상태이긴 하지만, 다른 조상에서 동등한 포화 상태를 달성하려면 추가 연구가 필요하다.